| ⎧ | Edge X = 131° 8' | |||
| ⎧ | Ρ ^ Ρ | ⎨ | ” Y = 82° 18' | |
| Angles measured | ⎨ | ⎩ | ” Z = 118° 40' | |
| ⎩ | Ρ ^ | α Ρ ὰ | = 114° 38' |
The pyramidal faces are generally etched so that the image is poor.
Title: On Sulphonfluoresceïn and Some of Its Derivatives
Author: C. W. Hayes
Release date: November 22, 2015 [eBook #50531]
Most recently updated: October 22, 2024
Language: English
Other information and formats: www.gutenberg.org/ebooks/50531
Credits: Produced by Paul Marshall and the Online Distributed
Proofreading Team at http://www.pgdp.net (This file was
produced from images generously made available by The
Internet Archive)
DISSERTATION
Presented for the Degree
of
Doctor of Philosophy
at the
Johns Hopkins University
by
C. Willard Hayes,
1887.
[Pg 2]
| p. | ||
| Introduction | 3. | |
| I | Ortho-sulpho-benzoic acid. | |
| Preparation from toluene and H2SO4 | 7. | |
| ” ” para-nitro-toluene and H2SO4 | 8. | |
| Oxidation of toluene - o - sodium sulphonate | 12. | |
| Preparation of Sulphinide from ” ” | 14. | |
| ” ” ” by chlorsulphonic acid. | 17. | |
| Conversion of sulphinide into o-sulphobenzoic acid. | 25. | |
| Analysis of and crystallography of ” ” ” | 28. | |
| II | Sulphonfluoresceïn—previous attempts | 29. |
| Preparation and purification of s-fluoresceïn | 30. | |
| Analyses of ” | 34. | |
| Properties of ” | 35. | |
| Salts. Ba. and Ca. of ” | 37. | |
| Acetyl derivative of ” | 41. | |
| Br substitution products of ” | 42. | |
| Action of H2SO4 on ” | 44. | |
| Action of HCl on ” | 45. | |
| Reduction of ” | 46. | |
| Conclusion. | 47. |
On Sulphonfluoresceïn and some of its Derivatives.
The close analogy in composition and structure between phthalic acid and ortho-sulpho-benzoic acid suggests the possibility of obtaining from the latter, by its action on phenols, substances analogous to the phthaleïns. If such compounds could be made they would afford a favorable opportunity of studying the effects produced in the properties of a compound by the replacement of CO by SO2. It was with a view to such study that the following discussed work was undertaken at the suggestion of Prof. Remsen and carried on under his constant guidance.
Some experiments previously performed by Remsen and Palmer (A.G.) [Pg 4] indicated the possibility of the formation of a fluorescent substance by the action of ortho-sulpho-benzoic acid on resorcin but they did not succeed in obtaining any definite crystallized compound from the reaction.
The chief obstacle to be overcome in the work is the difficulty in obtaining the o-sulpho-benzoic acid and a large proportion of the work here described was applied in that direction.
A method employed by Remsen and Fahlberg (Am. Ch. Jour. Vol. 1. p __ ) for getting the sulphonic acid group in the ortho position to [Pg 5] methyl was; (a) treat toluene with fuming H2SO4 forming thus ortho- and para- toluene sulphonic acids, (b) make the calcium salt of the sulphonic acids thus formed and from this the potassium salt. (c) treat this mixture of potassium toluene sulphonates with phosphorous penta-chloride forming the corresponding sulphonchlorides. One of these (para) being a solid and the other (ortho) an oily liquid a nearly complete separation could be effected. The difficulty with this method is however that the larger part of the product is the para and only a comparatively small proportion of the ortho compound is formed.
A second method employed consists in starting with p-nitro-toluene. This when treated with H2SO4 forms toluene p-nitro o-sulphonic acid. If now a method could be obtained for removing the nitro group the desired result would be attained. [Pg 6]
The attempt was made by Remsen and Palmer (A.G.) to accomplish this by (a) reducing the nitro compound to the amide, (b) making the diazo compound and (c) boiling this with absolute alcohol. According to generally accepted views this should effect the removal of the diazo group and its replacement by hydrogen.
Experiments however showed that the replacement was made not by hydrogen but by the ethoxy group -OC2H5. This method was therefore impracticable.
A modification of this method was suggested by an observation of Baeyer and Liebermann that if phenyl hydrazine be boiled with a dilute solution of copper sulphate the hydrazine group is replaced by hydrogen and benzene thus formed. Hence it was believed that if the hydrazine compound should be made from diazo compound mentioned above, the corresponding hydrocarbon, i.e. toluene o-sulphonic acid [Pg 7] could be obtained. The results of experiments showed that this afforded a practicable method of preparing toluene ortho-sulphonic acid.
After experimenting with various modifications of the method the following was found to be the best adapted to the purpose.
The potassium salt of toluene p-nitro-o-sulphonic acid is easily obtained, as already stated, by heating p-nitro toluene on the water bath with three times its weight of fuming H2SO4, neutralizing with chalk and to the solution of calcium salt thus obtained adding a slight excess of K2CO3. On filtering from the precipitated CaCO3 and evaporating slightly, the salt is obtained in long needle shaped crystals of a pale straw yellow color. This is
| ⎧ | CH3 | ||
| C6H3 | ⎨ | SO2OH | (o) |
| ⎩ | NO2 | (p). |
The reduction of the nitro group is best effected by means of tin and HCl, in the proportion, salt 5 parts, tin 6 parts and concentrated HCl 30 parts. [Pg 8]
The amido acid forms a compound with tin which crystallizes from the HCl together with stannous chloride. This compound may be broken up and the tin removed by continued boiling with water.
A better method of removing the tin is by dissolving the compound in Na2CO3. This forms a salt with the amido acid and throws down the tin as Sn(OH)2, a white flocculent precipitate. On filtering and adding to the solution conc. HCl, the free amido acid is deposited in characteristic colorless, rhombic crystals, having the formula
| ⎧ | CH3 | |
| C6H3 | ⎨ | SO2OH |
| ⎩ | NH2 |
The method at first employed for preparing the hydrazine compound consisted in treating the amido acid, suspended in HCl, with potassium nitrite and then with stannous chloride. The tin was then removed from the solution by the addition of sodium carbonate and the hydrazine compound thrown down with HCl. This method however gave [Pg 9] poor results the yield being only about 50% of the theoretical.
Another method was accordingly substituted for the above, namely that of Strecker and Römer (Ber. IV. s 784.) By this the diazo compound is made first and isolated. This is done by suspending the finely powdered acid in absolute alcohol, cooling and passing a current of the oxides of nitrogen through in the ordinary way. The acid changes in appearance, becoming more crystalline and slightly darker and settles quickly on being shaken. The reaction here may be expressed thus—
| ⎧ | CH3 | ⎧ | CH3 | ||
| C6H3 | ⎨ | SO2OH + HNO2 = | C6H3 | ⎨ | SO3 + 2H2O |
| ⎩ | NH2 | | | ╲ | ||
| ⎩ | N=N |
When the reaction is completed as shown by the appearance of the suspended powder it is filtered and while still fresh is added to a solution of acid sodium sulphite as long as it continues to dissolve readily. [Pg 10]
To this solution there is added a quantity of solution of acid sodium sulphite equivalent to that already used and the solution is then boiled. It has at first a deep red color but in a few moments becomes light reddish yellow. The reaction of HNaSO3 on the diazo compound may be represented in two stages, the first portion forming an addition product and the second acting as a reducing agent. Thus,
| ⎧ CH3 | ⎧ CH3 | |||
| 1. C6H3 | ⎨ SO3 + HNaSO3 = | C6H3 | ⎨ SO2ONa | |
| │ ╲ | ⎩ N=NSO3H | |||
| ⎩ N=N |
| ⎧ CH3 | ⎧ CH3 | ||
| 2. C6H3 | ⎨ SO2ONa + HNaSO3 + H2O = | C6H3 | ⎨ SO2ONa |
| ⎩ N=NSO3H | ⎩ NH—NHSO3 | ||
To the hot solution an excess of conc. HCl is added when the hydrazine compound separates in a few moments in lustrous yellow scales which completely fill the solution. On the addition of the HCl [Pg 11] a large amount of SO2 is given off from the excess of HNaSO3 and the solution becomes deep red. When the hydrazine has separated the mother liquor is again yellow.
The reaction is represented as follows:
| ⎧ CH3 | ⎧ CH3 | ||
| C6H3 | ⎨ SO2ONa + HCl + H2O = | C6H3 | ⎨ SO2OH + H2SO4 + NaCl |
| ⎩ NH—NHSO3H | ⎩ NH—NH2 |
The yield of hydrazine when both the diazo and the NaHSO3 are freshly prepared is practically quantitative.
The hydrazine thus prepared was treated with a hot 10% solution of copper sulphate till a permanent blue color was obtained in the solution. Nitrogen is evolved and the copper sulphate is reduced to cuprous oxide which is precipitated as a red powder. The reaction is as follows.
| ⎧ CH3 | ⎧ | ||
| C6H3 | ⎨ SO2OH + 2CuSO4 + H2O = | C6H4 | ⎨ CH3 + Cu2O + N2 + 2H2SO4 |
| ⎩ NH—NH2 | ⎩ SO2OH |
Chalk was added to the solution to precipitate the H2SO4 and form a calcium salt of toluene-o-sulphonic acid. From this the sodium salt was made by adding a slight excess of Na2SO3 and [Pg 12] evaporating to dryness. The salt is very soluble being deliquescent in the air while the corresponding potassium salt is not. From 1538 gr. of para-nitro-toluene, 655 gr. of toluene ortho-sodium sulphonate were obtained.
Having thus obtained the toluene ortho-sulphonic acid the next step in the problem was to find a convenient method for converting this into ortho-sulph-benzoic acid. Two ways present themselves for accomplishing this end. (1) direct oxidation of this salt and (2) conversion into benzoic sulphinide from which the acid may be obtained. Both of these methods were tried.
| ⎧ | CH3 | |
| C6H4 | ⎨ | |
| ⎩ | SO2ONa |
The sodium salt of toluene-o-sulphonic acid is oxidized to ortho-sulphobenzoic acid with considerable difficulty by KMnO4 in neutral solution. [Pg 13]
Thus two experiments showed that the oxidation was not complete after 24 hours boiling with excess of permanganate. If the solution be made alkaline however, the oxidation is completed in a few hours, yet the greatest difficulty still remains in the separation of the free acid from the products of oxidation in the solution. If HCl be added to the solution the acid salt
| COOH | ||
| ╱ | ||
| C6H4 | ||
| ╲ | ||
| SO2OK |
is formed and this has nearly the same solubility as the KCl also present. A better method therefore is to add a slight excess of H2SO4 and evaporate nearly to dryness. In this way are formed sulphates and the free acid presumably. The mixture is heated with alcohol (95%) which extracts the acid leaving the greater part of the manganese salts. This [Pg 14] extract is evaporated and reextracted with alcohol. To this solution BaCO3 is added to precipitate the H2SO4 and form the Barium salt of the o-sulphobenzoic acid. The solution is filtered from the BaSO4 and just enough H2SO4 is added to exactly precipitate the barium. The solution should thus contain only the free acid sought, which crystallizes out on evaporating to a small volume. While the method is theoretically possible it presents so many difficulties that it is practically useless. The yield is extremely small; only enough acid being obtained in this way to show that it was possible.
The second method for obtaining free o-sulphobenzoic acid from
toluene-ortho-sulphonic acid is by the conversion of the latter first
into benzoic sulphinide and then into the free acid. The sulphinide
was made essentially as described by Remsen (Am. Ch. Jour. Vol. I.
p. 428) with a few changes in the details as follows.
[Pg 15]
The salt
| ⎧ | CH3 | |
| C6H4 | ⎨ | |
| ⎩ | SO2ONa |
finely pulverized and in portions of from 10 to 50 gr. was placed in a Florence flask; an equivalent quantity of PCl5 added; An inverted condenser was then attached and the flask shaken. The action takes place at once and involves sufficient heat to distill off the oxychloride formed in the reaction. This being returned to the flask by the condenser furnishes a liquid medium in which the reaction takes place more readily and completely than when it is not present. It is best to cool the flask at first and afterwards heat gently on the water bath. The reaction which takes place may be represented as follows.
| ⎧ | CH3 | ⎧ | CH3 | ||||
| C6H4 | ⎨ | + PCl5 = | C6H4 | ⎨ | + POCl3 + NaCl. | ||
| ⎩ | SO2ONa | ⎩ | SO2Cl |
On the addition of water the chloride separates as a light yellow oil. This is washed with water and concentrated aqueous ammonia added, which forms toluene-o-sulphonamide thus. [Pg 16]
| CH3 | CH3 | ||||||
| ╱ | ╱ | ||||||
| C6H4 | + NH3 = | C6H4 | + HCl. | ||||
| ╲ | ╲ | ||||||
| SO2Cl | SO2NH2 |
The reaction is accompanied by a slight evolution of heat and the formation, apparently, of an intermediate product having a yellowish color, which passes over on longer standing into the white amide. After standing several hours the excess of ammonia was driven off by very gentle heating on the water bath. If the heat is too high a large amount of a tarry product is formed and the yield of amide is correspondingly small. In any case some of this tarry product is formed. When nearly all the ammonia had been driven off the mass was boiled with water which dissolves everything except the tar. The hot solution was filtered through charcoal and on cooling the amide separated in white feathery crystals which melt at 155°-156°.
The amide thus obtained was oxidized as described by Remsen (loc. cit.) [Pg 17] with potassium permanganate in neutral solution. The proportions are 10 gr amide. 40 gr KMnO4 and 1 L. water. The oxidation was usually effected in from four to six hours.
To obtain the sulphinide from this solution after oxidation, the latter, after filtration from the precipitated oxides of manganese, was slightly acidified with HCl and evaporated to about one fourth its original volume. On the addition of concentrated HCl to this solution, the sulphinide separated out in white or slightly yellowish feather shaped crystals melting at 212° and having the characteristic intensely sweet taste.
Before passing on to the methods used for converting the sulphinide into free acid another method should be described by which [Pg 18] the former was obtained in larger quantities and much more easily than by the one above described.
Beckurts and Otto (Ber. XI. 2061) found that by treatment of toluene with sulphuryl hydroxy-chloride or chlorsulphonic acid, ClSO2OH, both o- and p- and as they supposed also m-toluene sulphonchlorides were formed together with the corresponding sulphonic acid.
Claesson and Wallin (Ber. XII. p. 1848) repeated the work reaching practically the same results and finally Noyes (Am. Ch. Jour. Vol. VIII. p. 176) employed the reaction as a convenient method for obtaining toluene o-sulphon-chloride.
Chlorsulphonic acid is made by passing dry HCl over solid sulphuric acid so long as it continues to be absorbed. Since no solid sulphuric acid was at hand, ordinary fuming Nordhausen acid was taken and from [Pg 19] one of two equal portions the SO3 was driven over into the other. HCl was passed into the latter and the resulting chlorsulphonic acid distilled off at about 156°.
This was placed in a flask, provided with a drop funnel and exit tube, in portions of 150 gr. and to each portion 60 gr. of toluene was added, very slowly, with constant shaking, the temperature being kept near 10°. The action is violent and if any toluene is allowed to collect on the surface of the liquid it is apt to produce disastrous results. Large quantities of HCl are given off and the liquid in the flask assumes a brown color. When all the toluene has been added, it is poured into a large quantity of ice water, when the sulphon-chlorides separate out, the ortho- as a heavy oil and the para- as a white crystalline solid. After allowing to stand some time in order that as much of the para-chloride might crystallize as [Pg 20] possible the ortho- was drawn off and subjected to a freezing temperature for several hours. By this means more of the p-chloride was removed and the operation was replicated as long as any crystals continued to form, generally two or three times. In this way the greater part of the para- may be removed, though some still remains dissolved in the liquid chloride, which cannot be removed by repeated freezings.
The chloride thus obtained was treated with strong aqueous ammonia. The conversion to the sulphamide does not take place so readily as in case of the pure o-chloride obtained from the sulphonic acid and phosphorus pentachloride.
After standing about two days the whole of the oily chloride had solidified to a yellowish brown mass. The excess of ammonia was driven off by gentle heating on the water bath and the mass then boiled with water. Not enough water was added at first for complete [Pg 21] solution but when the first portion was saturated it was poured off through a filter and from it the amide separated in yellowish feathery crystals which melted at 105°-125° and consisted therefore as shown by Fahlberg (Am. Ch. Jour. Vol. I. p. 170) of a mixture of o- and p-sulphonamides. It was recrystallized and from it was obtained a portion melting at 153°-5° and one at 108°-20°.
Since this mixture cannot be completely separated by recrystallization another method was suggested. Remsen has shown that K2Cr2O7 in acid solution does not oxidize the methyl group in
| CH3 | ||
| ╱ | ||
| C6H4 | ||
| ╲ | ||
| SO2NH2 (o) |
but does oxidize that in
| CH3 | ||
| ╱ | ||
| C6H4 | ||
| ╲ | ||
| SO2NH2 (p). |
It was thought that in a mixture of the two the former might be left unchanged while the latter was oxidized to p-sulphamine benzoic acid.
To test this 15 gr of the mixture, melting at 105°-125°, was heated [Pg 22] with 40 gr K2Cr2O7, 55 gr H2SO4 and 2 vols. of water for about two hours. It was then tested and shown to be still a mixture of p-& o-amides, since it was again heated for several hours with half the original quantity of oxidizing mixture, then diluted, filtered and washed. The white crystalline residue was treated with sodium carbonate to dissolve the benzoic para-sulphamide and the residue was found to be pure toluene o-sulphonamide melting at 153°-155°. The small quantity remaining, 3 gr., indicated that part of the o-amide had been completely broken down by the strong oxidizing agent, though the proportion of o-& p-amides in the original mixtures was known only approximately. The evolution of gas during the oxidation would point to the same conclusion.
Although this effects a complete separation it is hardly economical [Pg 23] since it will be shown later that a separation can be conveniently effected after the oxidation with KMnO4 so that the o-amide contained in the mixture need not be lost.
The original mass was treated with successive portions of water till nothing remained but a black tarry substance. The amide which separated from these extracts was perfectly white and melted at 153-5°. It was therefore regarded as practically pure o-amide. The yield in amide melting above 153° was a little over one sixth the weight of toluene used.
The amide obtained in this way was oxidized in the manner already described. It was found however that there was always some benzoic p-sulphamide in the solution of the oxidation, due to the slight admixture of p- with the o-amide used. This is thrown down with the sulphinide on acidifying the solution and may be removed by [Pg 24] re-crystallization since it is somewhat less soluble in hot and cold water than sulphinide.
A better way to effect the separation, however, was found to be the following. After having evaporated the solution containing the products of oxidation, nearly neutralized with HCl, to about one fifth its original volume, it is made very slightly acid and allowed to cool. In this way very nearly all the benzoic p-sulphamide is separated from the solution and none of the sulphinide. After filtering, strong HCl is added and the sulphinide then separates in its characteristic form. This indicates that sulphinide forms an alkaline salt which is not decomposed by diluted HCl while the p-sulphamide does not.
The mixture of amides meeting at 105°-120° was oxidized and the products separated in this way gave about equal quantities of sulphinide and benzoic p-sulphamide. [Pg 25]
When toluene is treated with chlorsulphonic acid there are formed besides the ortho- and para- chlorides also ortho and para sulphonic acids. These of course are in solution in the water from which the chlorides separated. In order to recover the ortho-acid, the solution was neutralized with chalk forming the calcium salt: this converted into the potassium salt which by evaporating the solution to dryness was obtained as a white crystalline powder. When treated with PCl5 in the manner already described this gave a mixture of ortho and para sulphonchlorides consisting of about ⅓ ortho and ⅔ para.
Benzoic sulphinide may be converted into a sulpho-benzoic acid (1) by boiling with Ba(OH)2, (2) by heating in a closed tube with conc. HCl or (3) by evaporating on the water bath with HCl. [Pg 26]
1. Three gramms of sulphinide were boiled in a flask connected with an inverted condenser for about two days with an excess of Ba(OH)2. There was formed in the flask a hard mineral-like mass which was insoluble in water and cold diluted HCl but dissolved in hot HCl with effervesence. This was a Barium salt, probably basic (?) of ortho sulphobenzoic acid. There was also formed an easily soluble barium salt of that acid. The former was dissolved in H2SO4 and treated with BaCO3; the filtrate from the BaSO4 which contained a soluble barium salt was added to that above mentioned and the barium exactly precipitated with H2SO4 and the filtrate evaporated to dryness giving the free acid but not in a perfectly pure condition.
2. 2.75 gr. of sulphinide was sealed up in a tube with pure conc. HCl and heated two hours to 150°. On cooling nothing separated; the [Pg 27] liquid was evaporated to dryness giving 3.2 gr of acid and ammonium chloride. The reaction taking place here may be represented thus:
| CO | COOH | |||||||
| ╱ | ╲ | ╱ | ||||||
| C6H4 | NH + 2H2O + HCl = | C6H4 | + NH4Cl. | |||||
| ╲ | ╱ | ╲ | ||||||
| SO2 | SO2OH |
3. A more convenient method for obtaining the acid than either of the above, is to heat the sulphinide with conc. HCl on the water bath for two days. Then evaporate to dryness and dissolve the residue in a small quantity of water. If the sulphinide contained any para-sulphamide, as is usually the case, this will be left undissolved and most of the NH4Cl will crystallize on standing. This solution by slow evaporation deposits large colorless crystals of the free acid.
This acid is soluble in about two parts of cold water, very difficultly soluble in absolute alcohol and almost completely [Pg 28] insoluble in ether. It does not melt under 250° but considerably above that it melts, at first apparently without change and then with slight sublimation of a very deliquescent substance, probably the anhydride.
Two determinations of the S. made by Mr. A. F. Linn, gave the following results.
| I | ·1358 | gr substance gave | ·1855 | gr BaSO4 representing | 15·72% S. |
| II | ” | ” | ” | ” | ” |
Calculated for the formula
| COOH | |||
| ╱ | |||
| C6H4 | = 15·84% S. | ||
| ╲ | |||
| SO2OH |
|
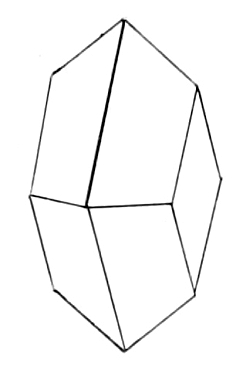
A crystallographic examination of the acid showed it to belong
to the orthorhombic system. Axial ratio: a: b: c = ·8507: 1: ·8121.
Planes. Ρ and α Ρ ὰ.
The pyramidal faces are generally etched so that the image is poor. |
Several attempts had already been made to obtain from the action of o-sulphobenzoic acid and resorcin a substance analogous to the fluoresceïn obtained by Baeyer[1] from phthalic anhydride and resorcin but while a strongly fluorescent substance was easily obtained, no definite compound could be separated from it. Thus Palmer obtained, by heating together the above named substances to 160°(?) a solid mass, part soluble in water and part insoluble as a dark brown amorphous powder. Both parts gave a strong fluorescence with alkalis. He was unable however to obtain the substance itself or any derivative in a crystalline form.
The first experiments in this series gave the same negative results. The mixture of acid and resorcin was heated in a sulphuric acid [Pg 30] both for several hours to 150°-170°, and as it showed no sign of solidification the temperature was raised to 200° and then to 235° where it was kept several hours longer. The black viscous mass obtained in this way became vitreous on cooling, and in all respects resembled that described by Palmer. This however is not the normal course of the reaction as shown later but is probably due to a decomposition of the normal product produced by too high heating.
An experiment was made with the acid salt of ortho-sulphobenzoic acid.
| COOH | ||
| ╱ | ||
| C6H4 | ||
| ╲ | ||
| SO2OK |
resorcin and H2SO4 heating the mixture to 150°-170°. A solid black mass was obtained strongly fluorescent in alkaline solution and in all other respects like the substance obtained above.
[Pg 31] As the result of a number of experiments the following method of preparing and purifying the sulphonfluoresceïn was found to give the best results.
The free acid is mixed with resorcin in the proportion of 1 part acid to 1.2 parts resorcin, or a slight excess over two molecules of the latter to one of the former. The mixture is placed in a deep vessel, a test tube or beaker, which is placed in a sulphuric acid bath, a thermometer being suspended in the mixture. The bath is heated and at about 100° the resorcin melts and the acid slowly dissolves in the liquid. No action appears to take place till the temperature reaches 175° where water begins to be given off and the liquid slowly assumes a darker color. White crystals of resorcin collect on the sides of the vessel. After the heating has been continued for about seven hours at 178°-185° the liquid has a clear deep red color but shows no [Pg 32] signs of becoming viscous. At length lustrous yellow crystalline plates appear in the liquid and soon the whole mass becomes a thick nearly solid yellow paste. Continuous heating at the same temperature causes no further apparent change. This mass, which on cooling is made up of yellow crystals imbedded in a red vitreous matrix, is then dissolved in hot water forming a clear red solution or at most one containing but a small quantity of a brown flocculent precipitate. This solution is filtered if necessary and evaporated to a small volume from which the substance separates on cooling in reddish yellow radial crystals. These are filtered and washed with ether till the washings are perfectly colorless. The substance on repeated crystallization from water has a pale straw yellow color and when deposited slowly forms transparent crystal blocks from 2·6 mm long, arranged in radial groups. [Pg 33]
A considerable amount of resorcin is lost by sublimation during the reaction especially if the operation is carried on in a beaker so that some excess should be added. But even when the resorcin is present in excess at the end of the reaction some free acid is always left which may be obtained from the mother liquid in the characteristic colorless orthorhombic crystals.
The water of crystallization and sulphur were determined in the new compound. In estimating the water, the substance was heated to 106°-123° for about ten hours till it attained a constant weight. On standing in the air it quickly regains its original weight. When heated to 130°-140° for some time it turns slightly reddish and loses over 10% of its weight which is not regained by standing in the air. [Pg 34]
Estimation No. I. was made by fusion with KOH and KNO3. Nos. II and III were made by Mr. Mindileff by Morse’s method, oxidizing with KMnO4 in KOH solution.
·3882 gr. heated to 106°-123° lost ·0302 gr. = 8·5%
Calculation for 2H2O - C19H12O6S + 2H2O = 8·9% water.
| I. | ·2007 | gr sub. gave | ·1286 | gr BaSO4 | = 8·77% S. |
| II. | gr ” ” | ” | gr ” | = 7·91” ” | |
| III. | gr ” ” | ” | gr ” | = 7·89” ” |
Calculation for C19H12O6S + 2H2O = 7·92% S.
These analyses show with but little doubt that the substance has the composition indicated above i.e. C19H12O6S + 2H2O. The reaction therefore which takes place between ortho-sulpho benzoic acid and resorcin from its analogy to that taking place between phthalic anhydride and resorcin may be represented thus, as shown by Baeyer in his second paper (Ann. 202. S. 43) [Pg 35]
Representing the formation of the anhydride as the first action.
| COOH | CO | |||||
| ╱ | ╱ | ╲ | ||||
| C6H4 | = C6H4 | O + H2O | ||||
| ╲ | ╲ | ╱ | ||||
| SO2OH (o) | SO2 |
and the action of resorcin on this anhydride thus.
| O | ||||||||||
| ╱ | ╲ | |||||||||
| (HO)H4C6 | C6H4(OH) | |||||||||
| ╲ | ╱ | |||||||||
| CO | OH | C | ||||||||
| ╱ | ╲ | ╱ | ╱ | ╲ | ||||||
| C6H4 | O + C6H4 | = | C6H4 | O + 2H2O. | ||||||
| ╲ | ╱ | ╲ | ╲ | ╱ | ||||||
| SO2 | OH (m) | SO2 |
The substance thus formed would naturally receive the name Sulphonfluoresceïn from its analogy with Fluoresceïn.
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sulphonfluoresceïn. | Fluoresceïn. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This compound shows a marked similarity to the fluoresceïn described by Baeyer as would naturally be expected from its great similarity of composition and constitution, but it also shows decided differences [Pg 36] which may be attributed to the replacement of CO by SO2.
Dissolved in water it shows a weak green fluorescence which in alkaline solution becomes much deeper but not by any means so strong as that of fluoresceïn. The dilute alkaline solution by transmitted light is almost perfectly colorless and by reflected light a clear green. Unlike fluoresceïn it is extremely soluble in water, about one part in two or three of hot and five or six of cold water. It is also soluble in absolute alcohol forming a yellow solution with weak fluorescence. It is soluble with difficulty in ether but when in solution is deposited only on evaporating to a small volume.
It does not melt at 250° but if held at a lower temperature for a long time becomes red undergoing some decomposition. If quickly heated somewhat above 300° it melts to a deep red liquid and then [Pg 37] solidifies. If the mass is treated with water it partly dissolves leaving a dark brown flocculent precipitate which dissolves on the addition of an alkali, the solution having an intense fluorescence, nearly if not quite equaling that of fluoresceïn. This change produced by heating was not further studied.
The crystals are very thin blades, apparently monoclinic, showing the clinopinacoid αΡὰ and a very narrow prism αΡ and clinodome Ρὰ. The angle β = 75° and the extinction angle against the ϲ axis = 20°. The axial ratio could not be accurately determined.
The influence of the SO2 group is shown by the fact that the substance acts as an acid decomposing carbonates and forming salts which is not the case with fluoresceïn. [Pg 38]
The substance was boiled with an excess of carefully purified BaCO3 for two hours. The filtrate from the BaCO3 evaporated to a small volume deposited yellow crystals resembling the original substance but in shorter and thicker prisms. These were twice recrystallized and had then a light straw yellow color.
A determination of the Ba gave the following results. The salt was dried in the air.
Transcriber's Note:
The following table was crossed out on the original. A note on the previous page beside the table was:
“All these calculations are wrong. J.R.”
| I | ·1078 | gr salt gave | ·0304 | gr BaSO4 | = 15·73% Ba. |
| II | ·1641 | ” ” ” | ·0457 | ” ” ” | = 15·53” ” |
| III | ·2425 | ” ” ” | ·0680 | ” ” ” | = 15·65” ” |
| IV | ·2860 | ” ” ” | ·0798 | ” ” ” | = 15·54” ” |
| V | ·1843 | ” ” ” | ·0498 | ” ” ” | = 15·08” ” |
| VI | ·2620 | ” ” ” | ·0708 | ” ” ” | = 15·08” ” |
| VII | ·3230 | ” ” ” | ·0906 | ” ” ” | = 15·65” ” |
| VIII | ·2875 | ” ” ” | ·0807 | ” ” ” | = 15·66” ” |
Calculated for C19H13O7SBa |
= 15·10% Ba. |
||||
[Pg 39] In the above determinations the salt analysed was taken from specimens made at three different times and purified in slightly different ways, Nos 1, 2, & 3 being washed with absolute alcohol. Nos V and V were made by precipitating the Ba with H2SO4 from a solution of the salt.
The water was determined by heating at 110° till constant weight was reached. Part only of the weight lost was regained on standing in the air.
·3943 gr salt lost at 110° ·0286 gr = 7.25%
Water calculated for C19H13O7SBa+2H2O = 7.35%
Although these analyses show a per cent. of Ba somewhat above that required by a compound having the formula C19H13O7SBa still this appears to be the most probable formula which can be assigned to the substance. If this is the true composition of the salt, then in [Pg 40] sulphonfluoresceïn the anhydride condition must be broken up by boiling with BaCO3 forming the salt thus.
| ⎧ | OH | ⎧ | OH | ||||||
| │ | ╱ | │ | ╱ | ||||||
| │ | C6H3 | │ | C6H3 | ||||||
| │ | ╲ | │ | ╲ | ||||||
| │ | O | │ | O | ||||||
| │ | ╱ | │ | ╱ | ||||||
| C | ⎨ | C6H3 | + 2H2O = | C | ⎨ | C6H3 | |||
| │ | ╲ | │ | ╲ | ||||||
| │ | OH | │ | OH | ||||||
| │ | C6H4SO2 | │ | C6H4SO2OH | ||||||
| │ | ╱ | │ | |||||||
| ⎩ | O | ⎩ | OH | ||||||
| ⎧ | OH | ⎧ | OH | ||||||||||
| │ | ╱ | │ | ╱ | ||||||||||
| │ | C6H3 | │ | C6H3 | ||||||||||
| │ | ╲ | │ | ╲ | ||||||||||
| │ | O | │ | O | ||||||||||
| │ | ╱ | │ | ╱ | ||||||||||
| 2 | C | ⎨ | C6H3 | + BaCO3 = | C | ⎨ | C6H3 | + Ba | |||||
| │ | ╲ | │ | ╲ | ||||||||||
| │ | OH | │ | OH | ||||||||||
| │ | C6H4SO2OH | │ | C6H4SO2OH | ||||||||||
| │ | │ | ||||||||||||
| ⎩ | OH | ⎩ | OH | 2 | |||||||||
By treating the salt with H2SO4 the original substance is reformed.
Attempts were made to prepare the calcium salt but without success. The S-fluoresceïn was boiled several hours with very finely powdered calcite, and some salt was formed as shown by the CO2 evolved but on evaporating the solution and recrystallizing the substance deposited it was found to be the unchanged S-fluoresceïn. Some Ca. salt was in the mother liquors but its extreme solubility prevented a separation being made. [Pg 41]
S.fluoresceïn was boiled with an excess of acetic anhydride for about three hours. The solution became quite dark and when evaporated on the water bath left a black tarry residue. This was treated with water which dissolved a part leaving a dark flocculent precipitate. The solution was boiled with animal charcoal and evaporated nearly to dryness. On cooling there separated a light yellow flocculent precipitate very soluble in hot water and but slightly less so in cold. This was dissolved in a small quantity of alcohol from which it separated on evaporation in small radial crystals having a light lavender color & satiny luster. They also have a peculiar odor resembling slippery elm which is not removed by recrystallization. [Pg 42] They show a tendency to decompose, becoming yellow on exposure to the air. The substance does not melt or change in appearance under 245°. With alkalis it gives a slight greenish fluorescence. From the method of its formation this was taken to be an acetyl derivative of S.fluoresceïn but whether the mono-or di-acetyl could not be determined without analysis for which the substance did not suffice.
It was especially interesting to see what influence the SO2 group would exert upon the introduction of Bromine into the compound. In the case of fluoresceïn four Bromine atoms enter easily and special precautions are necessary to obtain a product containing a smaller number. The case however is different with S.fluoresceïn.
The latter was dissolved in glacial acetic acid in which it is [Pg 43] soluble with some difficulty and to the solution was added a 20% solution of bromine in acetic acid, in sufficient quantity to make eight atoms of bromine to one molecule of S.fluoresceïn. This solution was evaporated on the water bath and while still having a considerable volume, small, red, sharply defined crystals began to separate. The solution was evaporated to a small volume and allowed to cool but nothing further separated. These crystals are difficultly soluble in water, alcohol and ether. The alkaline solution shows a green fluorescence and slight red color by transmitted light. These crystals were dissolved in a large quantity of alcohol which on evaporation gradually deposited very small yellow crystals, which were dried in the air and taken for analysis. The Br. was determined by Carius method.
| I. | ·2345 | gr sub. gave | ·1718 | gr AgBr | = 31·17% Br. |
| II. | ·2786 | ” ” ” | ·1815 | ” ” ” | = 27·72% Br. |
Calculated for C19H10Br2O6S |
= 30·42% Br. |
||||
[Pg 44] These results, though not conclusive, indicate that under the given conditions it is the di-bromsulphonfluoresceïn which is formed. Whether this is due to the presence in the compound of the SO2 group or simply to the greater insolubility of the di-than of the tetra-brom product cannot be definitely stated. When the original acetic acid mother liquor was evaporated to dryness, a red non-crystalline substance remained which more closely resembled rosin than the crystals. The concentrated alkaline solution had a deep red color without fluorescence and acted as a red dye stuff. The dilute alkaline solution showed the characteristic delicate pink of rosin.
A test tube in which S.fluoresceïn was being made just at the end of [Pg 45] the reaction broke and allowed the contents to run out into the sulphuric acid bath, which had a temperature of 175°. On standing several days the solution deposited a heavy precipitate which was separated by filtering through glass wool. When dry it formed a light yellow powder extremely soluble in water, alcohol and ether.
The alkaline solution had an intense green fluorescence with delicate shades of pink by transmitted light. On account of its great solubility it was impossible to purify it by crystallization, hence the Ba salt was made. The substance decomposed BaCO3 with great ease forming an easily soluble salt. When it was attempted to evaporate the solution of this salt to crystallization the latter came out in a hard insoluble granular form and on continuous boiling of the solution turned brown. To avoid this undesirable form it was [Pg 46] converted into the Ca. salt by treatment with H2SO4 and then CaCO3. This also formed a hard granular insoluble mass on boiling but did not change in color. As there was no guarantee as to its purity and only a small quantity was obtained it was not analyzed.
Hydrochloric acid does not dissolve S.fluoresceïn but converts it into a light yellow granular powder. When recrystallized from water in which it is quite easily soluble it melts partially at 130° apparently with some decomposition. This compound was not further studied.
When treated with zinc dust in a strong alkaline solution sulphonfluoresceïn is reduced to a colorless substance probably [Pg 47] analogous to fluoresceïn which is formed in the same manner. On account of its great solubility it could not be obtained in the free state. It is quickly oxidized to s.fluoresceïn by oxidizing agents as KMnO4 and HNO3 and passes back spontaneously on standing in the air. The latter action is however much slower than in case of fluoresceïn.
The principal results relating to s.fluoresceïn which have been reached in this work may be briefly summarized as follows. Orthosulphobenzoic acid acts on resorcin at a temperature of about 180° giving off water and forming a substance analogous to fluoresceïn but having the CO group replaced by SO2. This substance sulphonfluoresceïn crystallizes from water in light yellow monoclinic crystals having the composition C19H12O6S + 2H2O. It is very soluble in alcohol and water and with difficulty in ether. It [Pg 48] does not melt under 250° but above 300° melts with decomposition. It shows in alkaline solution a clear green fluorescence. It acts as an acid, decomposing carbonates and forming salts, the Ba salt having the composition C19H13O7SBa. It forms an acetyl compound when boiled with acetic anhydride. It forms substitution products with Br, probably the dibrom-product most easily. It forms a compound with H2SO4, probably a substitution product, whose composition was not determined. It is reduced by zinc dust and KOH to a colorless substance analogous to fluoresceïn.
Finally in terms of the prevalent theory the substance itself may be represented thus—
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Annalin. | No. | 183. | S. 1. No. 202. S. 36 & S. 153. |
| Berichte. | No. | IV. | S. 457. 555. 658. 662. |
| ” | ” | VIII. | S. 66. 146. |
Transcriber's Notes:
Illustration has been moved so it does not break up the paragraph.
As this was a hand-written thesis, the spelling, punctuation and hyphenation is very inconsistent.
The original spelling, hyphenation and punctuation have been left unchanged.