
Justus von Liebig
Title: History of Chemistry, Volume 2 (of 2)
Author: T. E. Thorpe
Release date: September 26, 2017 [eBook #55631]
Most recently updated: October 23, 2024
Language: English
Other information and formats: www.gutenberg.org/ebooks/55631
Credits: Produced by Sonya Schermann, Charlie Howard, and the Online
Distributed Proofreading Team at http://www.pgdp.net (This
file was produced from images generously made available
by The Internet Archive)
A HISTORY OF THE SCIENCES
Astronomy, by Prof. George Forbes, F.R.S.
Chemistry, 2 volumes, by Sir Edward Thorpe, C.B., D Sc., F.R.S., etc. (Director of Government Laboratories)
Old Testament Criticism, by Prof. Archibald Duff (Prof. of Hebrew and Old Testament Theology in the United College, Bradford)
In Active Preparation—
New Testament Criticism, by F. C. Conybeare, M.A. (Late Fellow and Prælector of Univ. Coll., Oxford)
Geology, by H. B. Woodward, F.R.S., F.G.S. (Assistant Director of Geological Survey)
Geography, by Dr. Scott Keltie, F.R.G.S., F.S.A.
G. P. PUTNAM’S SONS
New York London
A HISTORY OF THE SCIENCES
BY
Sir EDWARD THORPE
C.B., LL.D., F.R.S.
AUTHOR OF “ESSAYS IN HISTORICAL CHEMISTRY,” “HUMPHRY DAVY:
POET AND PHILOSOPHER,” “JOSEPH PRIESTLEY,” ETC., ETC.
TWO VOLUMES
II.
From 1850 to 1910
WITH ILLUSTRATIONS
G. P. PUTNAM’S SONS
NEW YORK AND LONDON
The Knickerbocker Press
1910
Copyright, 1910 by
G. P. PUTNAM’S SONS
This series is published in London by
The Rationalist Press Association, Limited
The Knickerbocker Press, New York
A History of the Sciences has been planned to present for the information of the general public a historic record of the great divisions of science. Each volume is the work of a writer who is accepted as an authority on his own subject-matter. The books are not to be considered as primers, but present thoroughly digested information on the relations borne by each great division of science to the changes in human ideas and to the intellectual development of mankind. The monographs explain how the principal scientific discoveries have been arrived at and the names of the workers to whom such discoveries are due.
The books will comprise each about 200 pages. Each volume will contain from 12 to 16 illustrations, including portraits of the discoverers and explanatory views and diagrams. Each volume contains also a concise but comprehensive bibliography of the subject-matter. The following volumes will be issued during the course of the autumn of 1909.
The History of Astronomy.
By George Forbes, M.A., F.R.S., M. Inst. C.E.; author of The Transit of Venus, etc.
The History of Chemistry: Vol. I. circa 2000 B.C. to 1850 A.D. Vol. II. 1850 A.D. to date.
By Sir Edward Thorpe, C.B., LL.D., F.R.S., Director of the Government Laboratories, London; Professor-elect and Director of the Chemical Laboratories of the Imperial College of Science and Technology; author of A Dictionary of Applied Chemistry.
To be followed by:
The History of Geography.
By Dr. John Scott Keltie, F.R.G.S., F.S.S., F.S.A., Hon. Mem. Geographical Societies of Paris, Berlin, Rome, Brussels, Amsterdam, Geneva, etc.; author of Report on Geographical Education, Applied Geography.
The History of Geology.
By Horace B. Woodward, F.R.S., F.G.S., Assistant Director of Geological Survey of England and Wales; author of The Geology of England and Wales, etc.
The History of Anthropology.
By A. C. Haddon, M.A., Sc.D., F.R.S., Lecturer in Ethnology, Cambridge and London; author of Study of Man, Magic and Fetishism, etc.
The History of Old Testament Criticism.
By Archibald Duff, Professor of Hebrew and Old Testament Theology in the United College, Bradford; author of Theology and Ethics of the Hebrews, Modern Old Testament Theology, etc.
The History of New Testament Criticism.
By F. C. Conybeare, M.A., late Fellow and Praelector of Univ. Coll., Oxford; Fellow of the British Academy; Doctor of Theology, honoris causa, of Giessen; Officer d’ Academie; author of Old Armenian Texts of Revelation, etc.
Further volumes are in plan on the following subjects:
Mathematics and Mechanics.
Molecular Physics, Heat, Life, and Electricity.
Human Physiology, Embryology, and Heredity.
Acoustics, Harmonics, and the Physiology of Hearing, together with Optics Chromatics, and Physiology of Seeing.
Psychology, Analytic, Comparative, and Experimental.
Sociology and Economics.
Ethics.
Comparative Philology.
Criticism, Historical Research, and Legends.
Comparative Mythology and the Science of Religions.
The Criticism of Ecclesiastical Institutions.
Culture, Moral and Intellectual, as Reflected in Imaginative Literature and in the Fine Arts.
Logic.
Philosophy.
Education.
v
| CHAPTER I | |
| PAGE | |
| State of Chemistry in the Middle of the Nineteenth Century | 1 |
| Introductory. Some Founders of Modern Chemistry: Liebig, Wöhler, Dumas. Rapid Extension of Organic Chemistry after 1850: Laurent and Gerhardt, Hofmann. Development of theory in Organic Chemistry. Other representative men of the middle period of the Nineteenth Century: Graham, Williamson, Bunsen. Modern Chemistry in relation to the Atomic Theory. | |
| CHAPTER II | |
| The Chemical Elements Discovered since 1850 | 26 |
| Nomenclature and Classification of Elements. Numerical relationships. Modes of discovery. The Spectroscope. Cæsium, rubidium, thallium, indium, gallium, scandium, germanium. The rare earth elements. Industrial applications of rare elements. | |
| CHAPTER III | |
| The Inactive Elements: Radium and Radio-Activity | 43 |
| Argon, helium, krypton, neon, and xenon. Radium. Disintegration theory of Rutherford and Soddy. Actinium and polonium. The emanations. | |
| CHAPTER IV | |
| Atoms and Molecules: Atomic Weights and Equivalents | 61 |
| Hypothesis of Avogadro. Stanislao Cannizzaro. viDetermination of molecular weights. Applicability of law of Dulong and Petit. Relations of molecular weight to osmotic pressures. Determination of atomic weights. Hypothesis of Prout, Dumas, Stas, Lord Rayleigh, Leduc, Morley, Guye, Theodore Richards. Validity of law of conservation of mass: Landolt. | |
| CHAPTER V | |
| The Molecular Theory of Gases | 79 |
| Interdependence of the gaseous laws. Kinetic theory of gases: Bernoulli, Waterston, Clausius, Maxwell, Boltzmann, Schmidt, Graham. Gaseous diffusion. Van der Waals’s equation. Ratio of specific heats: Kundt and Warburg. Liquefaction of gases. Critical temperatures and pressures: Andrews, Pictet and Cailletet, Wroblewski, Olszewki, Dewar, Kammerlingh Onnes. Liquefaction of air on the large scale. Research at low temperatures. | |
| CHAPTER VI | |
| The Periodic Law | 101 |
| Prout, Thomson, Döbereiner, Newlands, De Chancourtois. Statement of the Periodic Law by Mendeléeff and Lothar Meyer. Its importance as a system of classification. | |
| CHAPTER VII | |
| Valency | 112 |
| Origin of conception of Valency: Williamson, Gerhardt, Frankland, Couper, Kekulé. Tetravalency of carbon and linkage of atoms. Rational and constitutional formulæ. Dynamical theories of valency. | |
| CHAPTER VIII | |
| The Chemistry of Aromatic Compounds | 119 |
| Peculiarities of aromatic compounds. Kekulé’s benzene theory. Its applications. The essential oils. Terpenes. Camphor. Synthesis of viiperfumes. Alkaloids. | |
| CHAPTER IX | |
| Stereo-Isomerism: Stereo-Chemistry | 138 |
| Opticity: Biot, Mitscherlich, Pasteur, Wislicenus, Van ’t Hoff, Le Bel. Asymmetry. Racemisation. Multirotation. Geometrical isomerism. Geometrical inversion. Stereo-isomerism among nitrogen, sulphur, selenium, tin, and silicon compounds. Tautomerism. Steric hindrance. | |
| CHAPTER X | |
| Organic Synthesis: Condensation: Synthesis of Vital Products | 152 |
| Use of specific condensing reagents. Carbon suboxide. Artificial preparation of naturally occurring substances. Synthetic medicines. The ptomaines. Artificial alizarin. Indigo. The sugars and proteins: Emil Fischer. The doctrine of “vital force.” | |
| CHAPTER XI | |
| On the Development of Physical Chemistry Since 1850 | 171 |
| Molecular volumes of liquids. Nature of solution. Van ’t Hoff’s application of the gas laws to phenomena of solution. Osmosis and osmotic pressure. Traube. Pfeffer. Semi-permeable membranes. Measurement of osmotic pressure. Arrhenius. Doctrine of ionisation. Its applicability to the explanation of chemical phenomena. Thermo-chemistry. Mass action. Nature of reversible reactions. Thermal and Electrolytic dissociation. Relation between chemical nature and opticity, magnetic rotation and viscosity. Theory of phases. Catalysis. Enzyme action. Relations between valency and volume. Photochemistry. | |
| Bibliography | 187 |
| Index | 191 |
ix
| PAGE | ||
| Justus von Liebig | Frontispiece | |
| Jean Baptiste André Dumas | 9 | |
| Thomas Graham | 13 | |
| From a painting by G. F. Watts, R.A., in the possession of the Royal Society | ||
| Alexander William Williamson | 15 | |
| Bunsen, Kirchhoff, and Roscoe | 19 | |
| Sir William Ramsay | 48 | |
| Marie Curie (née Sklodowska) | 56 | |
| Stanislao Cannizzaro | 65 | |
| Sir James Dewar | 98 | |
| Dmitri Ivanowitsch Mendeléeff | 110 | |
| August Kekulé von Stradonitz | 126 | |
| Jacobus Henricus Van ’t Hoff | 142 | |
| Emil Fischer | 166 | |
| Svante August Arrhenius | 180 | |
1
In the preceding volume an attempt was made to outline the significant features in the development of chemistry, as an art and as a science, from the earliest times down to about the middle of the last century. Since that time chemistry has progressed at a rate and to an extent unparalleled at any period of its history. Not only have the number and variety of chemical products—inorganic and organic—been enormously increased, but the study of their modes of origin, properties, and relations has greatly extended our means of gaining an insight into the internal structure and constitution of bodies. This extraordinary development has carried the science beyond the limits of its own special field of inquiry, and has influenced every department2 of natural knowledge. Concurrently there has been a no less striking extension of its applications to the prosperity and material welfare of mankind.
With the death of Davy the era of brilliant discovery in chemistry, wrote Edward Turner, appeared for the moment to have terminated. Although the number of workers in the science steadily increased, the output of chemical literature in England actually diminished for some years; and, as regards inorganic chemistry, few first-rate discoveries were made during the two decades prior to 1850. Chemists seemed to be of Turner’s opinion that the time had arrived for reviewing their stock of information, and for submitting the principal facts and fundamental doctrines to the severest scrutiny. Their activity was employed not so much in searching for new compounds or new elements as in examining those already discovered. The foundations of the atomic theory were being securely laid. The ratios in which the elements of known compounds are united were being more exactly ascertained. The efforts of workers, Graham excepted, seemed to be spent more on points of detail, on the filling-in of little gaps in the chemical structure, as it then existed, than in attempts at new developments. For a time—during the early ’thirties—chemists struggled with the claims of rival methods of notation,3 and it was only gradually that the system of Berzelius gained general acceptance. At none of the British universities was there anything in the nature of practical tuition in chemistry. Thomson, at Glasgow, occasionally permitted a student to work under him, but no systematic instruction was ever attempted. The first impulses came from Graham in 1837, when he took charge of the chemical teaching at the University of London, and when, in 1841, he assisted to create the Chemical Society of London. Four years later the Royal College of Chemistry in London was founded and placed under the direction of August Wilhelm Hofmann—one of the most distinguished pupils of Liebig. Under his inspiration the study of practical chemistry made extraordinary progress, and discovery succeeded discovery in rapid succession. In bringing Hofmann to England we had, in fact, imported something of the spirit and power of his master, Liebig.
Among the pupils and co-workers of Hofmann were Warren de la Rue, Abel, Nicholson, Mansfield, Medlock, Crookes, Church, Griess, Martius, Sell, Divers, and Perkin. Whilst at Giessen he had begun the study of the organic bases in coal-tar with a view more especially of establishing the identity of Fritzsche’s anilin with the benzidam of Zinin and the krystallin of Unverdorben. Hofmann continued to cultivate4 with unremitting zeal the field thus entered. With Muspratt he discovered paratoluidine and nitraniline; with Cahours allyl alcohol. His pupil Mansfield worked out, at the cost of his life, the methods for the technical extraction of benzene and toluene from coal-tar, and thereby made the coal-tar colour-industry possible. It was in attempting to synthesise quinine by the oxidation of aniline that Perkin, then an assistant at the college, obtained, in 1856, aniline purple, or mauve, as it came to be called by the French, the first of the so-called coal-tar colouring matters. In 1859 this was followed by the discovery of magenta, or fuchsine, by Verquin. For its manufacture Medlock, one of Hofmann’s pupils, in 1860 devised a process by which for a time it was almost exclusively made. Hofmann studied the products thus obtained, and showed that they were derivatives of a base he called rosaniline; and he demonstrated that the colouring matters were only produced through the concurrent presence of aniline and toluidine. He also proved that the base of the dye, known as aniline blue, was triphenylrosaniline. As the result of these inquiries he obtained the violet or purple colouring matters known by his name. Lastly, all his classical work on the amines, ammonium compounds, and the analogous phosphorus derivatives was done at the Royal College of Chemistry.
5 Prior to the establishment by Liebig, in 1826, of the Giessen laboratory, the state of chemistry in Germany was not much, if at all, better than with us. The creation of the Giessen school initiated a movement which has culminated in the pre-eminent position which Germany now occupies in the chemical world. Students from every civilised country came to study and to work under its leader, and to carry away with them the influence of his example, the inspiration of his genius, and the stimulating power of his enthusiasm.
Justus von Liebig, was born at Darmstadt on May 12, 1803, and after graduating at Erlangen, where he worked on the fulminates, he repaired to Paris and entered the laboratory of Gay Lussac, with whom he continued his inquiries. Returning to Germany, he was appointed Professor of Chemistry at Giessen in 1826, and began those remarkable series of scientific contributions upon which the superstructure of organic chemistry largely rests. He investigated the cyanates, cyanides, ferrocyanides, thiocyanates, and their derivatives. In conjunction with Wöhler he discovered the group of the benzoic compounds and created the radical theory. With Wöhler also he investigated uric acid and its derivatives. He discovered hippuric acid, fulminuric acid, chloral, chloroform, aldehyde, thialdine, benzil, and elucidated the constitution6 of the organic acids and the amides. He greatly improved the methods of organic analysis, and was thereby enabled to determine the empirical formulæ of a number of carbon compounds of which the composition was imperfectly known. He practically laid the foundations of modern agricultural chemistry, and to his teaching is due the establishment of an important branch of technology—the manufacture of chemical fertilisers. He worked on physiological chemistry, especially on the elaboration of fat, on the nature of blood, bile, and on the juice of flesh. He studied the processes of fermentation, and of the decay of organised matter. He was a most prolific writer. The Royal Society’s Catalogue of Scientific Papers enumerates no fewer than 317 contributions from his pen. He was the founder of the Annalen der Chemie, which is now associated with his name, and of the Jahresbericht; he published an Encyclopædia of Pure and Applied Chemistry and a Handbook of Organic Chemistry. His Familiar Letters on Chemistry was translated into every modern language, and exercised a powerful influence in developing popular appreciation of the value and utility of science. Liebig left Giessen in 1852 to become Professor of Chemistry at the University of Munich and President of the Academy of Sciences. He died at Munich on April 18, 1874.
7 With the name of Liebig that of Wöhler is indissolubly connected. Although the greater part of their work was not published in conjunction, what they did together exercised a profound influence on the development of chemical theory.
Friedrich Wöhler was born at Eschersheim, near Frankfort, on July 31, 1800. After studying at Marburg, where he discovered, independently of Davy, cyanogen iodide, and worked on mercuric thiocyanate, he went to Heidelberg and investigated cyanic acid and its compounds, under the direction of Gmelin. In 1823 he worked with Berzelius at Stockholm, where he prepared some new tungsten compounds and practised mineral analysis. In 1825 he became a teacher of chemistry in the Berlin Trade School. Here he succeeded for the first time in preparing the metal aluminium and in effecting the synthesis of urea—one of the first organic compounds to be prepared from inorganic materials. Jointly with Liebig he worked upon mellitic and cyanic and cyanuric acids. In 1832 Wöhler, now appointed to the Polytechnic at Cassel, began with Liebig their memorable investigation on bitter-almond oil. In 1836 he was called to the Chair of Chemistry in the University of Göttingen, and with Liebig attacked the constitution of uric acid and its derivatives—the last great investigation the8 friends did in common. Wöhler subsequently devoted himself mainly to inorganic chemistry. He isolated crystalline boron, and prepared its nitrides, discovered the spontaneously inflammable silicon hydride, titanium nitride, and analysed great numbers of minerals and meteorites and compounds of the rarer metals. He made Göttingen famous as a school of chemistry. At the time of the one and twentieth year of his connection with the University it was found that upwards of 8000 students had listened to his lectures or worked in his laboratory. He died on September 23, 1882.
In France, Dumas exercised a no less powerful influence. If Liebig could reckon among his pupils Redtenbacher, Bromeis, Varrentrapp, Gregory, Playfair, Williamson, Gilbert, Brodie, Anderson, Gladstone, Hofmann, Will, and Fresenius; Dumas could point to Boullay, Piria, Stas, Melsens, Wurtz, and Leblanc—all of whom did yeoman service in developing the rapidly expanding branch of organic chemistry.

Jean Baptiste André Dumas was born on July 14, 1800, at Alais, where he was apprenticed to an apothecary. In his sixteenth year he went to Geneva and entered the pharmaceutical laboratory of Le Royer. Without, apparently, having received any systematic instruction in chemistry, he commenced the work of investigation. With Coindet he established the therapeutic910 value of iodine in the treatment of goître; with Prevost he attempted to isolate the active principle of digitalis, and studied the chemical changes in the development of the chick in the egg. In his twenty-fourth year Dumas went to Paris and became Répétiteur de Chimie at the École Polytechnique. He joined Audouin and Brongniart in founding the Annales des Sciences Naturelles, and began his great work on Chemistry Applied to the Arts, of which the first volume appeared in 1828. At about this time he devised his method of determining vapour densities, and published the results of a number of estimations made by means of it. With Boullay he began an inquiry on the compound ethers, out of which grew the etherin theory, which served as a stepping-stone to the theory of compound radicals—subsequently elaborated by Liebig and Wöhler. Dumas discovered the nature of oxamide and of ethyl oxamate, isolated methyl alcohol, and established the generic connection of groups of similarly constituted organic substances, or, in a word, the doctrine of homology. His work on the metaleptic action of chlorine upon organic substances eventually effected the overthrow of the electro-chemical theory of Berzelius and led to the theory of types, which, in the hands of Williamson, Laurent, Gerhardt, and Odling, was of great service in explaining the analogies and relationships of whole groups of11 organic compounds. He worked in every field of chemistry. He invented many analytical processes, established the gravimetric composition of water and of air, and revised the atomic weights of the greater number of the elements then known. Dumas exercised great influence in scientific and academic circles in France. He was an admirable speaker, and had rare literary gifts. On the creation of the Empire he was made a Senator, and was elected a member of the Municipal Council of Paris, of which he became president in 1859. He died on April 11, 1884.
It was largely through the influence of these master-minds that chemistry took a new departure. Prior to their time organic chemistry hardly existed as a branch of science: organic products, as a rule, were interesting only to the pharmacist mainly by reason of their technical or medicinal importance. But by the middle of the nineteenth century the richness of this hitherto untilled field became manifest, and scores of workers hastened to sow and to reap in it. The most striking feature, indeed, of the history of chemistry during the past sixty years has been the extraordinary expansion of the organic section of the science. The chemical literature relating to the compounds of carbon now exceeds in volume that devoted to all the rest of the elements.
12 In the middle of the nineteenth century chemists began to concern themselves with the systematisation of the results of the study of organic compounds, and something like a theory of organic chemistry gradually took shape. From this period we may date the attempts at expressing the internal nature, constitution, and relations of substances which, step by step, have culminated in our present representations of the structure and spatial arrangement of molecules. In 1850 the dualistic conceptions of Berzelius ceased to influence the doctrines of organic chemistry. The enunciation by Dumas of the principle of substitution, and its logical outcome in the nucleus theory and in the theory of types, had not only effected the overthrow of dualism, but was undermining the position of the radical theory of Liebig and Wöhler. The teaching of Gerhardt and Laurent had spread over Europe, and was influencing those younger chemists who, while renouncing dualism, were not wholly satisfied with a belief in compound radicals. Williamson’s discovery, in 1850, of the true nature of ether and of its relation to alcohol, and his subsequent preparation of mixed ethers, served not only to reconcile conflicting interpretations of the process of etherification, but also to reconcile the theory of types with that of radicals. Lastly, his method of representing the constitution of the1314 ethers and their mode of origin gave a powerful stimulus to the use of type-formulæ in expressing the nature and relations of organic compounds.

Thomas Graham.
From a painting by G. F. Watts, R.A., in the possession of the Royal Society.
Other representative men of the middle period of the nineteenth century, in addition to Williamson, were Graham and Bunsen. The three men were investigators of very different type, and their work had little in common. But each was identified with discoveries of a fundamental character, constituting turning-points in the history of chemical progress, valuable either as regards their bearing on chemical doctrine or as regards their influence on operative chemistry.
Thomas Graham was born in Glasgow on December 21, 1805, and, after studying under Thomas Thomson at the University of that city, attended the lectures of Hope and Leslie in Edinburgh. In 1830 he succeeded Ure as teacher of chemistry at Anderson’s College in Glasgow, and in 1837 was called to the Chair of Chemistry in the newly-founded University of London, in succession to Edward Turner. In 1854 he was made Master of the Mint. He died in London on September 16, 1869.
Graham’s work was mainly devoted to that section of the science now known as physical chemistry. His contributions to pure chemistry are few in number. By far the most important is his discovery of metaphosphoric acid and its relations to the other modifications of phosphoric1516 acid. Ortho- or ordinary phosphoric acid was known to Boyle; pyrophosphoric acid was discovered by Clark. Graham’s work is noteworthy as first definitely indicating the inherent property of the acids to combine with variable but definite amounts of basic substances by successive replacement of hydroxyl groups—the property we now term basicity, and was of fundamental importance in regard to its bearing on the constitution of acids and salts.
Graham’s fame chiefly rests upon his discovery of the law of gaseous diffusion (1829–1831), upon his work on the diffusion of liquids, and upon his recognition of the condensed form of hydrogen he termed hydrogenium. Questions involving the conception of molecular mobility, indeed, constituted the main feature of his inquiries. We owe to him, among others, the terms crystalloid, colloid, dialysis, atmolysis, occlusion—all of which have taken a permanent place in the terminology of science.

Alexander William Williamson was born at Wandsworth, London, on May 1, 1824. His father, a Scotchman and a fellow-clerk of James Mill (the father of John Stuart Mill) in the East India House, took an active share in the foundation, in 1826, of the University of London, subsequently known as University College. In 1840 the younger Williamson entered the University of Heidelberg with the intention of17 studying medicine; but, under the influence of Leopold Gmelin, he turned to chemistry. In 1844 he went to Giessen, to work under Liebig, and there made his first contributions to chemical science—viz., on the decomposition of oxides and salts by chlorine; on ozone; and on the blue compounds of cyanogen and iron. After graduating at Giessen he went, in 1846, to Paris, where he came under the influence of Comte, with whom he studied mathematics. In 1850, at Graham’s solicitation, he was appointed to the Chair of Practical Chemistry at University College, vacant by the death of Fownes. He at once embarked upon those researches which constitute his main contribution to science. In the attempt to build up the homologous series of the aliphatic alcohols from ordinary alcohol he succeeded in demonstrating the real nature of ether and its genetic relation to alcohol, and in explaining the process of etherification. The memoirs (1850–52) in which he embodied the facts had an immediate influence on the development of chemical theory. His explanation of the process of etherification familiarised chemists with the idea of the essentially dynamical nature of chemical change. He imported the conception of molecular mobility not only into the explanation of such metathetical reactions as the formation of the ethers, but into the interpretation of the phenomena of chemical change in general.18 In these papers, as also in one on the constitution of salts, published in 1851, he attempted to systematise the representation of the constitution and relations of oxidised substances—organic and inorganic—by showing how they may be regarded as built up upon the type of water considered as
in which the hydrogen atoms are replaced, wholly or in part, by other chemically equivalent atoms. This idea was immediately adopted by Gerhardt, was further elaborated by Odling and Kekulé, and was eventually developed into a theory of chemistry.
Williamson continued to direct the laboratory of University College until 1887, when he retired to the country. He died at Hindhead on May 6, 1904.
Robert Wilhelm Bunsen was born at Göttingen on March 31, 1811, and after studying chemistry under Stromeyer, the discoverer of cadmium, went to Paris and worked with Gay Lussac. In 1836 he succeeded Wöhler as teacher of chemistry in the Polytechnic School of Cassel, and in 1842 became Professor of Chemistry in the University of Marburg. In 1852 he was called to Heidelberg, and occupied the Chair of Chemistry there until his retirement in 1889. He died at Heidelberg on August 16, 1899.

Bunsen first distinguished himself by his1920 classical work on the cacodyl compounds, obtained as the result of an inquiry into the nature of the so-called “fuming liquor of Cadet,” an evil-smelling, highly poisonous, inflammable liquid formed by heating arsenious oxide with an alkaline acetate. The investigation (1837–1845) is noteworthy, not only for the skill it exhibits in dealing with a difficult and highly dangerous manipulative problem, but also for the remarkable nature of its results and on account of their influence on contemporary chemical theory. The research, in the words of Berzelius, was the foundation-stone of the theory of compound radicals. The name cacodyl or kakodyl was suggested by Berzelius in allusion to the nauseous smell of the compounds of the new radical arsinedimethyl, As (CH3)2, as it was subsequently termed by Kolbe.
Bunsen greatly improved the methods of gasometric analysis; these he applied, in conjunction with Playfair, to an examination of the gaseous products of the blast furnace in the manufacture of iron, and thereby demonstrated the enormous waste of energy occasioned by allowing the gases to escape unused into the air, as was then the universal practice. This inquiry effected a revolution in the manufacture of iron as important, indeed, as that due to the introduction of the hot blast.
Bunsen devised methods for determining the21 solubility of gases in liquids, for ascertaining the specific gravity of gases, their rates of diffusion, and of combination or inflammation. In 1841 he invented the carbon-zinc battery, and applied it to the electrolytic production of metals, notably of magnesium, the properties of which he first accurately described. In 1844 he contrived the grease-spot photo-meter, which was long in general use for ascertaining the photometric value of illuminating gas. His methods of ascertaining the specific heats of solids and liquids were simple, ingenious, and accurate. In 1855–1863 he carried out, in conjunction with Roscoe, a long series of investigations on the chemical action of light. In 1859, in association with Kirchhoff, he devised the first methods of spectrum analysis, and explained the origin and significance of the Fraunhofer lines in the solar spectrum, thus laying the foundations of solar and stellar chemistry. The application of the spectroscope to analytical chemistry almost immediately resulted in his discovery of cæsium and rubidium.
Bunsen worked on problems of chemical geology, and made a long series of analyses of volcanic products. With Schischkoff, he examined, in 1857, the products of fired gunpowder. He effected many improvements in analytical chemistry; devised the iodiometric method of volumetric analysis, and systematised22 the processes of water analysis. Lastly, he invented the gas-burner—a piece of apparatus with which his name is inseparably associated, and which has been of inestimable service to operative chemistry and in the arts. Bunsen was no theorist, and purely speculative questions had little or no interest for him. At the same time he was a great teacher, and made the chemical school of Heidelberg no less famous than the schools of Giessen and Göttingen.
The mass of material relating to the development of chemistry which has been accumulated during the past sixty years is so vast that it would be hopeless to attempt to survey it in detail within the limits of such a work as this. Nor, indeed, is this required in a history of this character. Those who desire information concerning the origin and sequence of the facts which collectively make up the superstructure of modern chemistry must be referred to the encyclopædias or larger treatises—or, preferably, to the numerous monographs, dealing with special sections, which the volume and complexity of the matter to be dealt with seem to render increasingly necessary. All we can do here is to attempt to show what has been the main outcome of this sixty years of incessant effort to elucidate the mysteries of chemical phenomena and to ascertain the nature of the conditions23 which control, modify, or determine them. All this effort is ultimately directed to the solution of the fundamental problem of the constitution of matter. The most significant result of this endeavour has been the elaboration and consolidation of the doctrine of chemical atoms, not necessarily of atoms in the limited Daltonian sense, but of atoms considered as associations of particles, or corpuscles—that is, of entities which may be divisible, but which, in the main, are not divided in the vast number of the transformations in which they are concerned. This modification of the original conception of Dalton has been thought by some to destroy the basis upon which his theory really rests. There is no necessity for such an assumption. So pronounced an atomist as Graham, as far back as 1863, in a suggestive paper entitled Speculative Ideas on the Constitution of Matter, enlarged the conception of the Daltonian atom in precisely the sense which recent experimental work appears to require. The present position, too, as it affects chemists, was equally well stated by Kekulé, in 1867, in the following terms:
The question whether atoms exist or not has but little significance from a chemical point; its discussion belongs rather to metaphysics. In chemistry we have only to decide whether the assumption of atoms is an hypothesis adapted to the explanation of24 chemical phenomena. More especially have we to consider the question whether a further development of the atomic hypothesis promises to advance our knowledge of the mechanism of chemical phenomena.
I have no hesitation in saying that, from a philosophical point of view, I do not believe in the actual existence of atoms, taking the word in its literal signification of indivisible particles of matter; I rather expect that we shall some day find for what we now call atoms a mathematico-mechanical explanation which will render an account of atomic weight, of atomicity, and of numerous other properties of the so-called atoms. As a chemist, however, I regard the assumption of atoms not only as advisable, but as absolutely necessary, in chemistry. I will even go further, and declare my belief that chemical atoms exist, provided the term be understood to denote those particles of matter which undergo no further division in chemical metamorphoses. Should the progress of science lead to a theory of the constitution of chemical atoms—important as such a knowledge might be for the general philosophy of matter—it would make but little alteration in chemistry itself. The chemical atoms will always remain the chemical unit; and for the specially chemical considerations we may always start from the constitution of atoms, and avail ourselves of the simplified expression thus obtained—that is to say, of the atomic hypothesis. We may, in fact, adopt the view of Dumas and of Faraday—that, whether matter be atomic or not, thus much is25 certain: that, granting it to be atomic, it would appear as it now does.1
1 The Study of Chemical Composition, by Ida Freund (Cambridge University Press), 1904.
The greater part of that which follows will be devoted, therefore, to an exposition of certain of the great advances in knowledge—many of them of primary importance—which have been made during the last fifty or sixty years and which have served to strengthen this extended conception of the atomic theory, and to establish its position as an article of the scientific faith of the twentieth century.
26
In 1850 the number of substances generally recognised as chemical elements, in the sense in which that term was first employed by Boyle, was sixty-two. Two members—viz., the pelopium of Rose and the ilmenium of Hermann—were, however, subsequently shown to be identical with metals already known. At the present time (1910) the number of the chemical elements definitely recognised as such is eighty-two. In 1850, as now, they were broadly classified as metals and non-metals, although it was felt then, no less strongly than now, that no very clear line of demarcation was traceable between the two groups. Sixty years ago the elements usually styled non-metals were thirteen in number; to-day the number is nineteen—the increase being due to the inclusion of arsenic and the discovery of the so-called inactive elements, helium, argon, krypton, neon, and xenon. In 1850 there were forty-seven elements definitely27 classed as metals; in 1910 the number is sixty-three.
At all periods in the history of chemistry as a science the general tendency has been to name substances, whenever possible, in accordance with the theoretical conceptions of the time, and hence it has happened that the same body at successive periods has had very dissimilar names. But in naming the substances we term elements, theoretical conceptions are not usually applicable. Oxygen, it is true, derives its name from such a conception; and, etymologically, the name connotes an error. Hydrogen, too, has no more right to be called the water former than oxygen. Davy, who invented the term chlorine, advocated that the chemical elements should be named from some distinguishing peculiarity, either of origin or of physical property. In the main this principle has been adopted especially in later years although there are numerous instances of names derived from pure arbitrary sources. It is largely for the reason that the names of the elements are, with rare exceptions, unconnected with theories that they have remained unchanged, whereas names of compounds, which are far more frequently dependent upon speculative ideas, have constantly been altered in order to comply with the prevailing hypotheses of the period. At the same time it is not always clear that the etymology28 of certain of the elements is well ascertained. It has been recently shown, for example that the commonly accepted origin of the word “antimony” from antimoine, based on the alleged experiences of mediæval ecclesiastics has no valid foundation. The word is, in reality, derived from the Arabic alhmoud: this became latinised to althimodium and eventually to antimonium.
By the middle of the nineteenth century the system of symbolical notation suggested by Berzelius was everywhere current; and, stripped largely of its dualistic associations, this system still remains the most generally convenient method of expressing the composition, analogies, and numerical relations of substances. During the middle of the last century philosophic chemists, although subscribing, with hardly an exception, to the doctrine of definite combining proportions, were by no means agreed as to the sufficiency of Dalton’s explanation of the experimental laws of chemical combination; and the hypothesis of atoms in the Daltonian sense was not universally accepted. To some the atomic theory of Dalton, which assumed that the combining proportion was identical with the relative weight of the atom, was unnecessary as an explanation of the laws of combination. Or at most it was only one out of a variety of molecular conditions in which matter might29 exist. Consequently some chemists were in the habit of drawing a distinction between chemical atoms and physical atoms. The chemical atom was identical with the Daltonian atom but this was by no means the same as the physical atom of Democritus or Leucippus. The view in 1850, in fact, was not very dissimilar from that to which recent experimental inquiry has led. But it can hardly be said that the doubts were dependent upon valid experimental evidence; they arose rather from the erroneous interpretation of imperfectly ascertained facts—upon the supposed inconsistencies of the law of Gay Lussac with the hypotheses of Avogadro and Ampère. As soon as the facts were clearly perceived and the inconsistencies reconciled we heard less of the supposed distinction between the chemical and the physical atom. It is only within quite recent time, and as the result of entirely new lines of inquiry, that the distinction has been revived.
In the early part of the last century attempts were made by Berzelius to classify the chemical elements according to their electro-chemical relations, and by Thomson according as they were “supporters” or “non-supporters of combustion.” It was soon perceived that Thomson’s system had no philosophical basis, and it quickly fell into disuse. After the discovery of isomorphism, an endeavour was made by Graham to arrange the simple bodies in accordance30 with their natural relations, and even before 1850 the various elements were grouped by him very much as now.
This scheme of classification, somewhat modified by considerations of valency, and occasionally corrected by more accurate information concerning true analogies (as when vanadium was transferred by Roscoe to the nitrogen group), was in general use for practically a quarter of a century—in fact, until it was superseded by the gradual adoption of Mendeléeff’s arrangement based on periodicity. There can, however, be little doubt that this attempt by Graham at a natural classification paved the way along which Newlands and eventually Mendeléeff were led to devise our present rational system of grouping the chemical elements.
The numerical relationships existing among the equivalents and atomic weights of the elements of certain of these groups, pointed out by Dumas, Pettenkofer, Odling, Gladstone, and others, gave rise to much speculation. The values of the gradational differences, of course, depended upon whether equivalents or atomic weights were employed; but the immediate point is that, whichever basis was adopted, definite numerical relations were to be perceived. Thus, in the case of the group of the halogens, it was pointed out that the individual members are connected together as follows:
31
| Fluorine. | Chlorine. | Bromine. | Iodine. | |
|---|---|---|---|---|
| 19 | 35.5 | 80 | 127 | |
| a | a + d | a + 2d + d´ | 2a + 2d + 2d´ |
where a = 19; d = 16.5; d´ = 28.
Thus, too, in the case of the nitrogen group:
| Nitrogen. | Phosphorus. | Arsenic. | Antimony. | Bismuth. | |
|---|---|---|---|---|---|
| 14 | 31 | 75 | 119 | 207 | |
| a | a + d | a + d + d´ | a + d + 2d´ | a + d + 4d´ |
where a = 14; d = 17; d´ = 44.
On the basis of these and similar numerical relationships it was surmised that, just as the successive members of a group of homologous organic radicals are formed by increments of CH2, so the substances in the several groups of the elements may be produced by successive additions of some form of matter common to them all. This has its counterpart, somewhat modified, in the modern hypothesis of the disintegration of the elements. Dumas conceived the elements in any particular group to be built up by successive accretions of particular forms of matter; Rutherford and Soddy suppose them to be derived by the successive elimination of matter from some unstable parent substance.
Since 1850 the existence of at least twenty-two new elements may be said to have been established. Of course, many more than this number have been announced, more or less tentatively;32 but subsequent investigation has either not confirmed their existence, or has definitely disproved it. The names, symbols, and atomic weights of the twenty-two, arranged in alphabetical order, are as follows:
| Argon | A | 39.9 | |
| Cæsium | Cs | 132.8 | |
| Dysprosium | Dy | 162.5 | |
| Europium | Eu | 152.0 | |
| Gadolinium | Gd | 157.3 | |
| Gallium | Ga | 69.9 | |
| Germanium | Ge | 72.5 | |
| Helium | He | 4.0 | |
| Indium | In | 114.8 | |
| Krypton | Kr | 83.0 | |
| Lutecium | Lu | 174.0 | |
| Neodymium | Nd | 144.3 | |
| Neon | Ne | 20.0 | |
| Praseodymium | Pr | 140.6 | |
| Radium | Ra | 226.4 | |
| Rubidium | Rb | 85.4 | |
| Samarium | Sa | 150.4 | |
| Scandium | Sc | 44.1 | |
| Thallium | Tl | 204.0 | |
| Thulium | Tm | 168.5 | |
| Xenon | Xe | 130.7 | |
| Ytterbium (Neoytterbium) |
} | Yb | 172.0 |
The additions have been due, to some extent, to the refinement of processes of analysis already in use, but more especially to the employment of new analytical methods; or, lastly, to the application of a generalisation concerning the mutual relations of the elements which has served to indicate not only the existence of new and specific members of families of elements already known, but to point out the probable mode of their occurrence.2
2 The substances which appear to be formed by the disintegration of uranium, radium, thorium—the so-called radio-active elements—such as ionium, actinium, polonium, and the various emanations to which they give rise, are not here enumerated. They are dealt with in Chapter III.
33
Although the existence of the element fluorine was surmised as far back as 1771, when Scheele first recognised that the product of the action of oil of vitriol upon fluor-spar contained a hitherto unknown substance, it was not until 1886 that this substance was definitely isolated by Moissan by the electrolysis of the acid potassium fluoride in solution in hydrogen fluoride. Cerium tetrafluoride, CeF4, and lead tetrafluoride, PbF4, when heated, were observed by Brauner to evolve a gas having a smell resembling that of hypochlorous acid, which was probably free fluorine. Certain violet-coloured varieties of fluor-spar, when powdered, emit a peculiar smell, which has been attributed to free fluorine.
Gore observed that anhydrous hydrogen fluoride would not conduct electricity—a fact confirmed by Moissan. Moissan found, however, that on adding potassium fluoride to the liquid it readily suffered electrolysis with the liberation of free fluorine as a light greenish yellow gas with a pungent, irritating smell resembling that of hypochlorous acid. It has a vapour density corresponding with an atomic weight 19. By the application of cold and pressure it may be liquefied. At still lower temperatures34 it may be frozen to a white solid. Fluorine is characterised by an extraordinary chemical activity, and combines, even at ordinary temperatures, with a large number of substances. Sulphur, phosphorus, arsenic, antimony, boron, iodine, and silicon inflame or become incandescent in contact with it. It combines with hydrogen with explosive violence, even in the dark and at the lowest temperature. It unites also with the metals, occasionally with incandescence, and decomposes water with liberation of oxygen.
The application, by Bunsen, of the spectroscope to chemical analysis almost immediately resulted in his discovery, in 1860, of cæsium, and, in 1861, of rubidium. Cæsium was first detected in the mineral water of Dürkheim in the Palatinate and in the mineral petalite, by the two blue lines it forms in the spectrum, whence its name from the Latin cæsius, used to designate the blue of the clear sky. Rubidium was found in a lepidolite by means of a number of lines in different parts of the spectrum not previously observed, two being especially remarkable in the outermost region of the visible red portion—whence the name of the element from the Latin rubidus, used to designate the darkest red colour. The new metals were found to have the closest analogies to potassium, with which they usually occur associated in nature.35 Rubidium is found in a number of lepidolites, leucite, spodumene, triphylite, mica, and orthoclase, and in the Stassfurt carnallite; in sea-water and in many mineral waters. It occurs also in the ashes of many plants such as those of beetroot, tobacco, tea, coffee, etc. It is doubtful if it is a normal constituent of plant food, attempts to introduce it in place of potash having failed. It is not improbable that these elements would have remained unknown except for spectrum analysis. At all events, one of them—cæsium—was missed in 1846 by Plattner, in the course of the analysis of the mineral pollucite, in which it occurs to the extent of one third of its weight. After the discovery of cæsium by Bunsen, this mineral was again analysed by Pisani, when it was found that the alkali which Plattner had mistaken for potassium was in reality cæsium. Cæsium is found to a very small extent in many mineral waters, in a variety of minerals, and in the ashes of plants.
In 1861 Sir William Crookes made known the existence of a new element which he called thallium. He found it in a seleniferous deposit obtained from an oil of vitriol factory in the Harz. It was characterised by giving a bright green line in the spectroscope—whence its name from θαλλός, a green or budding twig. The discovery was confirmed in the following year by Lamy. Thallium, in its general chemical relations,36 has many analogies to the metals of the alkalis although in the metallic state it has the closest resemblance to lead. It occurs in many varieties of pyrites, in a few minerals, such as crookesite, lorandite, zinc-blende and copper pyrites, etc., and in certain mineral waters.
In 1863 Reich and Richter, by means of the spectroscope, detected the presence of a new element in the zinc-blende of Freiberg. The observation that it afforded two indigo-blue lines in the spark-spectrum led them to give it the name indium. It has since been found in numerous blendes, in various zinc and tungsten ores, and in many iron ores. It is a silver-white, ductile, and malleable metal, melting at 174°, and burning when heated with a violet flame. It is related in chemical characters to aluminium and zinc. Its true place in the natural scheme of classification of the elements was indicated by Mendeléeff.
In 1875 Lecoq de Boisbaudran discovered a new element in the zinc-blende of Pierrefitte in the Pyrenees, also by means of spectrum analysis. The spark-spectrum of its salts affords two characteristic violet lines quite different in position from those given by indium. To the new element its discoverer gave the name of gallium. It has been found in very small amounts in other blendes, but is still one of the rarest of the chemical elements. It is a bluish-37white, hard, and slightly malleable metal fusing at a temperature not much higher than that of a hot summer day. Its existence and main properties, as well as its more significant chemical relationships, were predicted by Mendeléeff in 1869 from considerations based upon his periodic law. (See ante.)
In the same year Mendeléeff also predicted the existence of a new element belonging to the group of which boron is the first member, which he provisionally termed eka-boron, and described its main properties. Mendeléeff’s prediction was verified in 1879 by Nilson’s discovery of the element scandium. Scandium occurs associated with yttrium, ytterbium, etc., in many Swedish minerals, such as euxenite, gadolinite, yttrotitanite, etc. The metal itself has not been isolated, but the properties of its compounds correspond closely with those of the corresponding ekaboron compounds, as predicted by Mendeléeff.
A further illustration of the value of the principle of periodicity, as developed by Mendeléeff, in indicating the existence of new elements, is seen in the discovery of germanium. In 1885 Weisbach discovered a new Freiberg silver mineral, to which he gave the name argyrodite. This on analysis by Winkler was found to contain a new element to the extent of about seven per cent. with properties identical with those predicted by Mendeléeff for a missing element38 in the fourth group of the periodic series, consisting of silicon, tin, and lead, and which he had provisionally termed eka-silicon. Argyrodite, in fact, is a double sulphide of silver and germanium, 2Ag2S.GeS2. Germanium is a greyish-white, lustrous metal of sp.gr. 5.5., melting at about 900°, and resembling silicon and tin in its general chemical relations.
Dysprosium, europium, gadolinium, lutecium, neodymium, praseodymium, samarium, thulium, and ytterbium (neoytterbium) belong, like scandium, to the group of the so-called rare earth metals. These substances have been detected in a great variety of minerals, many of which are extremely rare. The elements most frequently occur in nature associated with yttrium, cerium, thorium, and zirconium.
Dysprosium was first detected, in 1886, by Lecoq de Boisbaudran in the so-called erbium earth of Mosander, in which Cleve had previously (1880) announced the existence of two other elements, holmium and thulium. There is some reason to believe that the holmium of Cleve is identical with dysprosium. Ytterbium was discovered by Marignac, in 1878, in the mineral gadolinite. In 1906 Auer von Welsbach announced that Marignac’s ytterbia was a mixture, which was confirmed in the following year by Urbain, who separated it into two elements, which he named neoytterbium and lutecium.39 Europium was discovered by Demarçay in 1901. All these earths are met with in small quantities associated with yttria in gadolinite, euxenite, samarskite, xenotime, cerite, orthite, and other similar minerals. Their compounds, or such of them as have been described, resemble the corresponding compounds of yttria. They are recognised by differences in their spectroscopic behaviour. Gadolinium was detected, independently, in 1886, by Marignac and Lecoq de Boisbaudran in the terbium earth of Mosander.
What was long known as didymium (διδυμος = a twin) was discovered by Mosander in 1841. It owes its name to its close chemical relationship to, and almost constant association with, lanthanum—both elements occurring in many minerals, more particularly in cerite, allanite, and monazite. In 1885 Auer von Welsbach announced that the didymium of Mosander was, in reality, a mixture of two elements which could be separated by the systematic fractional crystallisation of the double ammonium nitrates; to these elements he gave the names praseodymium (πράσινος, leek-green) and neodymium (νέος, new). Neodymium salts are rose-coloured, whereas those of praseodymium are green, and the elements are further characterised by differences in their absorption and spark-spectra. When mixed, the substances give the spectrum40 originally considered to be characteristic of didymium.
Samarium was discovered in 1879 by Lecoq de Boisbaudran in samarskite. Its salts are yellow, and afford in solution characteristic absorption bands.
It is not improbable that many of the minerals from which the so-called rare earths are obtained contain elements hitherto unrecognised, and it is possible that certain of the substances now assumed to be elements may, like didymium, turn out to be mixtures. In fact, additional elements have from time to time been announced, as for example, the decipium of Delafontaine (1878) and the monium or victorium of Crookes (1899), pronounced by Urbain to be identical with gadolinium: their individuality cannot as yet be said to be established. Didymium itself was stated by Krüss and Nilson (1888) to be even more complicated than the work of Auer von Welsbach would seem to indicate, and to contain no fewer than eight elementary substances. As yet, however, no confirmation of this surmise has been obtained.
The chemistry of the rare earths has of late years been greatly extended owing to the employment of certain of the members of the group in the manufacture of the “mantles” used in gas-lighting, and which consist substantially of thoria, mixed with about one per cent. of ceria.41 Large quantities of monazite, thorianite, thorite, cerite, and other minerals, are now worked up for the sake of the thoria and ceria they contain, and considerable amounts of residual products, consisting largely of other members of the family, are now available for investigation. It is reasonably certain, therefore, that our knowledge of this section of inorganic chemistry will be largely augmented in the immediate future. Indeed, the application of thoria to the construction of gas-mantles may be said to have removed that substance from the category of the rare elements. No sooner was it discovered that it was capable of useful application than unexpected sources of supply were found.
The same result has followed in other cases. One of the most significant developments of modern chemistry is seen in the efforts which are constantly being made to turn the so-called rare elements to useful account; and when they are found to be technically valuable it is generally observed that hitherto unknown sources of supply are soon available. Cerium salts have been found to be useful in the colouring of glass and porcelain, as mordants in dyeing, in photography, and in medicine. Zirconium has been used in incandescent electric lighting, and thallium has been employed in the manufacture of highly refractive optical glass. Titanium, molybdenum, and vanadium are used in the42 manufacture of steel of high tensile strength. Tantalum and tungsten are employed in the construction of filaments in incandescent electric lighting. Tantalum, indeed, has been found to occur in considerable quantities, and to be more largely distributed than was hitherto supposed. Alloys of tungsten and aluminium are used in automobile construction, and alloys of tungsten, aluminium, and copper in the manufacture of propeller blades. Tungsten steel is used in armour plates, and to stiffen the springs of cars; in the manufacture of piano-wire, and to increase the permanency of magnets. Even the rarer metals of the platinum group are finding many important applications. Osmium-iridium is used for the bearings of compasses, for the tips of gold pens, and in the construction of standard weights. Osmium and ruthenium enter into the composition of filaments for electric lighting. The extraordinary influence of light on the electric conductivity of selenium has been made use of in the transmission of photographs by telegraph and telephone wires, and for measuring the light intensity of the Röntgen rays in clinical work.
43
Argon, helium, krypton, neon, and xenon belong to the group of the so-called inactive elements, and constitute what are known as the rare gases of the atmosphere. The existence of these bodies is of great theoretical value and few discoveries of recent times have exacted more interest and curiosity. Twenty years ago it was generally assumed that practically all that was to be known concerning the composition of atmospheric air had been ascertained. Priestley and Cavendish had recognised that it was mainly composed of oxygen and nitrogen, and Cavendish had definitely stated that these gases are present in practically constant proportion, independent of season, climate, or locality. Thénard, Saussure, and others, had determined the limits of variation in the amount of carbon dioxide. Bunsen and Regnault had established that the quantities of oxygen and nitrogen are subject to slight alteration, the44 extent of which could be readily determined by the exact eudiometric processes they had devised. Lastly, it was proved beyond a doubt that the gases of the atmosphere are simply mechanically mixed, and can be separated by a variety of physical methods. In fact, of no single subject could it be more confidently assumed that finality of knowledge had apparently been reached.
In 1892, in the course of a series of determinations of the densities of the common gases, Lord Rayleigh found that the density of nitrogen obtained from the air was slightly greater than the density of that gas prepared by the decomposition of ammonia and of nitric acid, the difference in weight being about 1 part in 200—an amount far greater than could be accounted for by errors of weighing. Various suppositions were made in explanation of the discrepancy; but these, when tested, were found not to account for the facts. By heating the atmospheric nitrogen with metallic magnesium, whereby the greater portion of the gas is absorbed to form the nitride, Sir William Ramsay found that the density of the residual gas was still further increased, which rendered it probable that the relatively high density of atmospheric nitrogen as compared with that derived from ammonia, and, as Lord Rayleigh found, from other sources also, was due to the presence of a gaseous substance45 in the air of considerably greater density than nitrogen or oxygen. Lord Rayleigh also subjected atmospheric nitrogen mixed with oxygen to the electric discharge over a solution of caustic soda, in a manner similar to that already employed by Cavendish, and found also that the residual gas was considerably increased in density. At the Oxford meeting of the British Association in August, 1894, the two investigators were in a position to announce that the discrepancy was actually due to the presence of a hitherto unknown gaseous constituent of atmospheric air, considerably more soluble in water than nitrogen, and to which, on account of its chemical inertness, the name of argon (ἀργον, idle) was given. By a special apparatus devised by Lord Rayleigh, in which a mixture of air and oxygen is submitted to an electric flame produced by a powerful, rapidly alternating current, considerable quantities of argon were separated from the air. It has also been found that by the use of metallic calcium or a mixture of magnesium and lime, the atmospheric nitrogen is absorbed at a lower temperature, and more rapidly than by magnesium alone.
Argon has been found to exist in the gases from springs and mineral waters, notably in those of Bath, Cauterets, Wildbad, and Harrogate. It has also been found in a meteorite,46 in the gas occluded in rock-salt, and in the minerals malacone, uraninite, brōggerite, etc. No animal or vegetable substance appears to contain it. It is present in atmospheric air to the extent of about one per cent. by volume. It is a colourless gas of an atomic weight of 39.9: one litre of it at the standard temperature and pressure weighs 1.7815 grams. Experiments made by the method of Kundt and Warburg—i.e., by determining the ratio of the specific heats at constant pressure and constant volume by the velocity of sound in the gas—prove that argon, like mercury gas, is monatomic. This of itself indicates that argon is an element, since a monatomic compound is a contradiction in terms. The calculations from the experimental data presuppose that argon obeys the laws of Boyle and Dalton, which was found on trial to be the case. By the application of cold and pressure argon can be liquefied. The liquid boils at -186°.1 and freezes at -187°.9. The spectrum of the gas is exceedingly complicated, consisting of a great number of lines extending throughout the visible portion and far into the extreme red and ultra-violet. The colour of the light emitted on sparking the gas changes with increase of temperature from a brilliant red to a bright blue—depending on the intensity of the discharge. All attempts to induce argon to enter into combination with47 other substances have failed. The methods of its preparation show that it does not combine with oxygen, although Troost and Ouvrard state that it unites with magnesium vapour. It forms no compounds with hydrogen, chlorine, phosphorus, sulphur, sodium, tellurium, etc. Even fluorine, probably the most generally active of the chemical elements, shows no tendency to unite with it.
In 1888 Dr. Hillebrand, of the U.S. Geological Survey, in examining a form of uraninite known as cleveite, so named from the late Professor Cleve, found that on treatment with dilute sulphuric acid it gave off considerable quantities of a gas which was assumed to consist only of nitrogen, as it gave the spectroscopic reactions of that element. To test whether this gas contained argon, Ramsay, in 1895, further examined it spectroscopically. After sparking it with oxygen in the presence of caustic soda solution, in the way already described, it gave no indications of argon. The main characteristic of its spectrum was a bright yellow line, known as D3, not coincident with that afforded by sodium, but identical in position with a line detected in the chromosphere during the solar eclipse of 1868, which line, on examination by Frankland and Lockyer, could not be ascribed to any known element. For this supposed new element the name helium, from ἥλιος, the sun,4849 had been suggested. This was the first occasion on which an element observed originally only in the sun was found to occur also on the earth. The presence of the new element in the gas from cleveite was subsequently confirmed by Langlet working in Cleve’s laboratory.

Helium is a monatomic gas having the atomic weight 4. It is less soluble in water than argon. Like argon, it shows no tendency to enter into chemical union with any other substance. It has been found in many minerals, particularly in those containing uranium and the so-called rare earth metals. It also occurs among the gases issuing from certain mineral springs, such as those of Bath and at Cauterets in the Pyrenees, and also at Adano near Padua. The spectrum of helium contains, in addition to the characteristic yellow line—by which its presence had been recognised not only in the solar chromosphere, but also in certain of the fixed stars—two lines in the red, and lines in the green, blue, and violet. The character of the light emitted by the spark-discharge is modified by the intensity of the discharge in a manner similar to that of argon. It has been shown by Collie that its spectrum is altered by the presence of mercury vapour. It is the least refractive of all the gases. Helium was liquefied by Kammerlingh Onnes in 1908. It forms a colourless liquid of sp. gr. 0.154, boiling at50 -268.5; that is, 4°.5 above the absolute zero of temperature. Its critical temperature is about 5° absolute, and its critical pressure above 2¼ atmospheres.
The methods now in use for obtaining liquid air, referred to in a subsequent chapter, enable large quantities of that material to be obtained readily; and it was in investigating spectroscopically the residues left after volatilising a quantity of liquid air that Ramsay and Travers, in 1898, detected the existence of two new monatomic gaseous constituents of the air which they named respectively krypton (χρυπτός, hidden) and neon (νέος, new), the former heavier and the latter lighter than argon. By fractional distillation of the argon, simultaneously procured, a gas was obtained which in the spectroscope showed the characteristic lines of helium—previously recognised in atmospheric argon by Kayser and Friedländer—together with a complicated spectrum consisting of a number of lines in the red, orange, and yellow due to the new element neon. On cooling this mixture to -252° by means of liquid hydrogen, the neon solidified, while the helium remained gaseous and could thus be separated.
Krypton was obtained from the residues left on the evaporation of a large quantity of liquid air. Mixed with the krypton was a third51 gaseous constituent of air, to which the name xenon (ξενος, the stranger) was given. The boiling-point of krypton at atmospheric pressure was found to be -152°, and its melting-point -169°; the boiling-point of xenon was -109° and its melting-point -140°. Their critical temperatures were respectively -62°.5 and +14°.7. Hence xenon could be liquefied by pressure a very little below the mean temperature of the air. Neon boils at -243° and freezes at -253°. They form colourless liquids freezing to ice-like solids. All of them, with the exception of argon, which is present to the extent of about 1 part in 107 parts of air, are contained in extremely small amounts in the atmosphere, approximately in the following proportions:
| Helium | 1 | part in | 245,300 | parts by volume. |
| Neon | 1 | ” ” | 80,800 | ” ” |
| Krypton | 1 | ” ” | 20 millions | ” ” |
| Xenon | 1 | ” ” | 170 ” | ” ” |
Many tons of liquefied air have since been systematically fractionated, but no other gas than those above named has been obtained.
Julius Thomsen, of Copenhagen, in a paper published in 1895, entitled On the Probability of the Existence of a Group of Inactive Elements, pointed out, in relation to Mendeléeff’s Law of Periodicity (see ante), that in periodic functions the change from negative to positive value, or52 the reverse, can take place only by a passage through zero or through infinity; in the first case the change is gradual, and in the second case it is sudden. The first case corresponds with the gradual change in electrical character with rising atomic weight in the separate series of the periodic system, and the second case corresponds with a passage from one series to the next. It therefore appears that the passage from one series to the next in the periodic system should take place through an element which is electrically indifferent. The valency of such an element would be zero, and therefore in this respect also it would represent a transitional stage in the passage from the univalent electronegative elements of the seventh to the univalent electropositive elements of the first group. This indicates the possible existence of a group of inactive elements with the atomic weights 4, 20, 36, 84, 132—numbers corresponding fairly closely with the atomic weights respectively of helium, neon, argon, krypton, and xenon.
No discovery of recent years has created more widespread interest than that of the radio-active elements.
In 1896 Henri Becquerel found that uranium salts emitted an invisible radiation which had the power of affecting a photographic plate,53 even though not directly exposed to it, exactly in the same way as the Röntgen or X-rays. Since that time a number of substances have been shown to possess a similar property. Such substances are said to be radio-active. The radiation emitted by them is not uniform in character. It has been found to be of three distinct types, known respectively as the α, β, and γ radiations. The α rays consist of positively electrified particles moving with a velocity equal to about a fifteenth of that of light. These rays have little penetrative power, and are capable of being deflected by a magnet.
The β rays consist of negatively electrified particles of a mass not greater than one thousandth of that of the hydrogen atom, and they move with a velocity approximating to that of light. The β rays have a greater penetrative power than the α rays, and are even more readily deflected by a magnet.
The γ rays are analogous to, if not identical with, the X or Röntgen rays; they move with the velocity of light, have a high penetrative power, but are not affected by the magnet. All three forms of radiation render gases electrically conductive, excite luminescence or fluorescence in certain substances, change the colour of glass, convert oxygen into ozone and yellow phosphorus into red phosphorus, and act upon photographic plates.
54 According to the disintegration theory of Rutherford and Soddy, the radio-active elements are forms of matter undergoing changes resulting in the formation of new forms possessing chemical and physical properties differing from those of the parent substance, these changes being accompanied by the production of sensible heat, or some other manifestation of energy, due to the process of transformation of the changing atoms. The rate of change is found to be different for each radio-active element, but to be constant for the same element irrespective of its particular form of combination. The relative radio-activity of the various chemical combinations of a given radio-active element is directly proportional to the quantity of the element contained in them. The process of disintegration may be carried through a number of intermediate products until a stable form is produced. Uranium, in which the phenomenon of radio-activity was first perceived, is supposed to give rise to no fewer than seventeen different forms of matter, including radium, actinium, and polonium. Thorium, another radio-active element, is supposed to disintegrate into eight different forms of matter. Uranium disintegrates with extreme slowness; it is calculated that in a year not more than one ten-billionth part of the uranium is transformed. The first disintegration product is termed uranium x.55 If a quantity of dehydrated uranium nitrate be treated with ordinary ether, a slight residue is obtained which is found to contain uranium x. It emits β and γ rays, and is relatively rapidly transformed into other substances. Ordinary uranium, freed from uranium x, only emits α rays. Uranium salts can be freed from uranium x by repeated crystallisation, uranium x remaining in the mother liquors.

The existence of radium was first made known by Mme. Curie in 1898. In examining certain uranium minerals and uranium products, Mme. Curie observed that their radio-activity was apparently greater than that corresponding with the amount of uranium contained in them, and she was led to surmise that this might be due to the presence of some constituent more strongly radio-active than uranium. This supposition proved to be well founded, and she eventually succeeded in isolating a new element termed radium, forming compounds with characters and relationships akin to those of barium. The richest source of radium at present known consists of certain residues occurring at Joachimsthal, in Bohemia, left after the extraction of uranium from pitch-blende, in which radium occurs to the extent of 0.2 gram per ton. These residues are mainly sulphates of lead and calcium, mixed with a great variety of other metallic compounds. To obtain the radium the mixture5657 is heated with concentrated caustic soda solution, the residue washed with water and treated with hydrochloric acid which dissolves the greater portion of the material. Nearly the whole of the radium is left in the insoluble portion. This, after washing with water, is boiled with a solution of sodium carbonate so as to transform the alkali-earths into carbonates. These are converted into chlorides or bromides from which, by repeated crystallisation, barium chloride or bromide is obtained, containing the greater portion of the radium as a halide salt. The radium and barium salts are then separated by fractional crystallisation, the radium salts being slightly less soluble in water and alcohol, and in solutions containing the halogen acid, than the barium salt.
Pure radium chloride (RaCl2) is a white crystalline salt, resembling barium chloride, with which it appears to be isomorphous. Radium, like barium, forms an insoluble carbonate and sulphate, but a soluble nitrate and bromide. The bromide is much less stable than the chloride; on standing it evolves bromine and becomes basic. Radium has as yet been obtained in such small quantities that very few of its compounds have been prepared.
The rays from radium salts burn the skin, and are found to be useful in the destruction of rodent ulcers; they appear to act upon proteids, destroy58 bacteria, bleach chlorophyll, and affect the germinative power of seeds. A pure and freshly-prepared salt of radium seems to emit only α rays, but it soon forms disintegration products, and then gives out, in addition, the β and γ rays.
In the process of disintegration the salts emit heat corresponding to about 75 gram calories per hour for each gram of radium present; their temperature is thus uniformly higher than that of their environment. One product of the change probably connected with the emission of the α rays, is the gas helium.
Radium has an atomic weight of 226.5. It is regarded as a product of the disintegration of uranium, the atomic weight of which is 238.5. It is believed to have been formed through an intermediate product known as ionium, a radio-active element discovered by Boltwood in the mineral carnolite. The atomic weight of ionium is surmised to be about 230. Radium itself is supposed to form at least eight disintegration products, the first of which is the so-called emanation, discovered by Dorn in 1900, an inactive gas with an atomic weight of about 180, giving a bright line spectrum, decomposing into helium, liberating oxygen and hydrogen from water, and capable of being condensed to a liquid and solidified at a low temperature. Ramsay and Gray have determined its physical59 constants. The liquid is phosphorescent and shines with a colour depending on the nature of the glass of the vessel which contains it. The solid is also phosphorescent, the colour varying with the temperature. It gives out only α rays and in its disintegration, like radium, evolves heat. Its position in the Periodic Table is probably above that of xenon. Other products are known as radio-lead and polonium. The latter substance was identified by M. and Mme. Curie in 1898, and was the first of the strongly radio-active substances to be recognised. In the periodic system it seems to follow bismuth and to be a member of Group VI., with a possible atomic weight of 210. Its spectroscopic characters have recently been examined by Mme. Curie and Debierne, who have shown that in its decay it evolves helium.
The rate of disintegration of radium is relatively slow; it has been calculated that the time required for half of any given quantity of radium to change completely into other products is about 2000 years. Rutherford has calculated that in 26,000 years a kilogram of radium would be reduced to one milligram of active substance, the remainder having passed into degradation products.
In 1899 Debierne announced the existence of another radio-active element contained in uranium minerals, which he termed actinium. This is probably a disintegration product of uranium60 and identical with the emanium of Giesel. It occurs associated with the rare earths which can be separated from the pitch-blende residues, and is eventually found in the lanthanum salts. Nothing is known as to its atomic weight or its chemical relationships. It undergoes change, and forms, apparently, a gaseous emanation which rapidly disintegrates and can be condensed to a liquid at a low temperature. Four other successive products have been identified by the character of the radiation they emit, their degradation constants, and the time required for one half of any given quantity to disintegrate into other forms of matter.
Thorium was shown to contain a radio-active element by Mme. Curie and Schmidt, independently, in 1898. Whether thorium is itself active is doubtful. The rate of disintegration of radio-thorium is probably greater than that of uranium. It, too, seems to form a gaseous emanation which can be condensed at the temperature of liquid air and appears to be an inert gas of high molecular weight with the characteristics of the argon family.
The type of radiation emitted by the several products has been observed, and their constants of change and half-value periods calculated; but little or nothing is known at present concerning their atomic weights, spectroscopic or chemical characters.
61
It has already been pointed out that the discovery by Gay Lussac, and independently by Dalton, that gases combine in simple proportions by volume, and that the volume of the gaseous product, measured under comparable conditions of temperature and pressure, stand in simple relation to the volumes of the constituents, seemed to most of Dalton’s contemporaries, but not to Dalton himself, to afford strong evidence of the validity of his explanation of the essential nature of chemical combination. It appeared obvious from the facts that there must exist some simple relation between the densities, or specific gravities, of the elementary gases and their atomic weights. When, however, the principle underlying Gay Lussac’s law was extended so as to include gases in general—both simple and compound—difficulties were met with which were only satisfactorily cleared away during the latter half of the nineteenth62 century. The first rational attempt to explain the facts observed by Dalton and Gay Lussac, concerning the volumetric relations of gases, was made in 1813 by Amedeo Avogadro by the assumption that a given volume of all gases—simple or compound—contains the same number of integral molecules; hence the relative weights of these volumes represent the relative weights of the molecules. According to Avogadro, in the case of the simple gases the integral molecules are composed of a certain number of elementary molecules of the same kind, whereas the integral molecules of compound gases and vapours are made up of elementary molecules of different kinds. The elementary molecule of Avogadro is now termed the atom; his integral molecule we call simply a molecule. Similar conceptions were published independently by Ampère in 1814. It follows from the doctrine of Avogadro and Ampère that, as the number of integral molecules is the same in equal volumes of all gases, these molecules must be equidistant from each other, their mutual distances depending upon pressure and temperature. This at once serves to explain the laws of Boyle and Dalton that gases, no matter what their chemical nature, behave identically, as regards change of volume, when compressed by pressure or expanded by heat.
The true significance of the hypotheses of63 Avogadro and Ampère was long obscured, first on account of their imperfect appreciation by the great leaders of chemical thought during the first half of the nineteenth century—Berzelius, Gay Lussac, Wollaston, and Gmelin—and, secondly, on account of the almost universal practice of deducing atomic weights from purely chemical considerations of equivalence. At the same time, it must be admitted that the recognition of the value of these hypotheses was still further retarded by the seeming anomalies which resulted from a more extended knowledge of the vapour densities of elements and compounds. Thus the vapour densities of mercury, sulphur, phosphorus, and arsenic, as ascertained by Dumas and Mitscherlich, were plainly inconsistent with chemical analogies and the law of Dulong and Petit. So, too, what appeared to be the vapour densities of sal-ammoniac, phosphorus pentachloride, sulphuric acid, calomel, and of other substances that might be mentioned, were not in accordance with the values demanded by other well-ascertained facts.
By the middle of the nineteenth century the hypothesis of Avogadro was practically forgotten and the law of volumes ignored. The atomic weights of the elements, and the system of notation universally employed in England and Germany, were based wholly upon equivalents.64 The anomalies thus created were clearly pointed out by Gerhardt, and subsequently by Laurent who showed how a consistent and harmonious explanation of the facts could be reached by regarding as true equivalents equal volumes—for example, of steam, ammonia, hydrogen chloride, carbon dioxide, marsh gas, etc.; by assuming, in other words, that equal numbers of the molecules of these various substances are contained in equal volumes of the gases, as contended by Avogadro and Ampère. The simplicity and consistency of the new notation gradually won for it the adhesion of chemists. This adhesion was facilitated by the memorable researches of Williamson on etherification, of Gerhardt on the anhydrides, and by the work of Frankland on the radicals; and it reached its logical conclusion when Cannizzaro—on the basis of Avogadro’s hypothesis—discussed, in 1858, the true atomic weights of the metallic elements as distinguished from their equivalent values. In certain of its aspects the new table of atomic weights drawn up by Cannizzaro resembled that originally proposed by Berzelius; but the numbers adopted by the Swedish chemist were founded on no uniform or rational basis, and were frequently inconsistent.

Our present tables of atomic weight bring, therefore, the values for the several elements into harmony with the doctrine of Avogadro and6566 Ampère, with the law of Dulong and Petit, and with the facts of isomorphism. The values are, in fact, in unison with all the criteria which serve to indicate the atomic weights of the elements. As a result, our present system of notation, which is, of course, based upon these atomic weights, assigns formulæ to compounds which indicate their true relative molecular weights and simplify the accurate expression of their relationships and chemical transformations. One by one the instances of anomalous vapour density, which were so many stumbling blocks to the universal acceptance of a system based upon the law of gaseous volumes, have been shown to be not only not inconsistent with it, but actually so many corroborative proofs. Thus, in the case of ammonium chloride, the observed vapour density of which was found to be practically half its calculated value, it has been proved that the vapour of this salt, when heated to the temperature at which the observations were made, is mainly resolved into molecules of ammonia and hydrogen chloride, which together occupy double the space of the ammonium chloride molecule. Phosphorus pentachloride vapour, on being sufficiently heated, is similarly more or less resolved into phosphorus trichloride and chlorine.
Moreover, it has been found, by a more accurate study of the action of heat upon the67 vapour of ammonium chloride and of phosphorus pentachloride, that within certain narrow limits of temperature these substances can actually exist as such in the gaseous state, and that the density of their vapours does actually conform to that demanded by theory. Moreover, phosphorus pentafluoride, the analogue of the pentachloride, is gaseous at ordinary temperatures, and has a normal density. It may be heated to a high temperature without showing any sign of decomposition.
Limitations of space will not allow of a fuller explanation of the apparent anomalies already alluded to. They have each in turn been experimentally attacked and satisfactorily explained. No valid exception is now known to the universal applicability of the principle.
To-day the chemical history of a substance, whether elementary or compound, if vaporisable, is not complete until its vapour density is known, since a knowledge of this constant affords the most certain means of establishing the relative weight of its molecule. Accordingly many chemists have endeavoured to simplify and render more convenient the modes of determining vapour densities. Thanks to the efforts of Hofmann and Victor Meyer, the processes associated with the names of Dumas, Gay Lussac, Deville, and Troost, which have furnished us with valuable information in the past,68 have now given way to comparatively simple and rapid methods, which, although not necessarily more accurate, furnish the required information with less expenditure of time and trouble; that is to say, they serve to indicate which of two, or more, presumed molecular weights is correct, and so enable us to establish the molecular formula of the substance. The chemical formula of a substance is a condensed expression of a number of facts connected with its history. Thus the expression H2O—the chemical formula for water—indicates that the substance is composed of hydrogen and oxygen, in the proportion, using round numbers, of 2 parts by weight of hydrogen and 16 parts by weight of oxygen; or, in other words, of 2 atoms of hydrogen, each weighing 1, and 1 atom of oxygen weighing 16. The formula, moreover, connotes the fact that when the gases combine 2 volumes of hydrogen unite with 1 volume of oxygen to form 2 volumes of water-vapour (steam). So, too, the formula HCl—which represents hydrogen chloride—means that the substance is a compound of 1 atom of hydrogen weighing 1 united with 1 atom of chlorine weighing 35.5; it also denotes the fact that in the act of union 1 volume of hydrogen combines with 1 volume of chlorine to form 2 volumes of hydrogen chloride. Lastly, the formula NH3 signifies that the molecule of ammonia is composed of69 1 atom of nitrogen weighing 14, and 3 atoms of hydrogen, each weighing 1; it further indicates that when ammonia gas is resolved into its constituents, as it can be when sufficiently heated, 2 volumes of ammonia gas are increased to 4 volumes of a mixture made up of 1 volume of nitrogen and 3 volumes of hydrogen.
In short, all the formulæ are what are called two-volume formulæ; that is, the relative molecular weights of the substances occupy the same volume as two relative parts by weight of hydrogen. Hence their vapour densities—referred to hydrogen as unity—are the halves respectively of their molecular weights. Steam is 9 times, hydrogen chloride 18.25 times, and ammonia 8.5 times heavier than hydrogen, when measured under identical conditions of temperature and pressure. The quantities expressed by the formulæ H2, H2O, HCl, NH3, occupy the same volume. This is equally true of the quantities expressed by O2, N2, Cl2, etc. These expressions signify that the molecules of the elements of hydrogen, oxygen, nitrogen, and chlorine consist each of 2 atoms of the respective substances: the molecule of water consists of 3 atoms—2 of hydrogen and 1 of oxygen; the molecule of hydrogen chloride of 2 atoms—1 of hydrogen and 1 of chlorine; whereas the molecule of ammonia contains 4 atoms—1 atom of nitrogen and 3 atoms of hydrogen.
70 Certain of the elementary bodies are, as already stated, capable of existing in different allotropic states. Thus there is a modification of oxygen known as ozone. This substance has long been recognised as being formed from air under the influence of the electric discharge. It was the subject of study by Schönbein as far back as 1839; but that it was a condensed form of oxygen and not a peroxide of hydrogen—as was at one time surmised—was first established by Andrews and Tait. The degree of its condensation was definitely ascertained by Soret by observing its rate of diffusion, from which, on the basis of Graham’s law, its density could be inferred. It was found to be 1½ times that of ordinary oxygen. Hence, if the molecule of oxygen consists of 2 atoms, that of ozone consists of 3 atoms. The chemical symbol of ozone is, therefore, O3.
It has been found also that the molecule of sulphur, in the state of solution, contains eight atoms. This complex molecule in the gaseous state gradually breaks down as the temperature is increased, and at temperatures above 850° contains, like its analogue oxygen, only two atoms. The molecules of phosphorus and arsenic, in the gaseous state, are each found to consist of four atoms. On the other hand, the molecules of mercury, zinc, and cadmium each consist of only one atom. As will be shown71 later, Kundt and Warburg established, in 1875, the fact that mercury vapour is a monatomic gas, by determining the rate at which sound is propagated through it. In the same way it was shown that helium, argon, and its congeners are also, as already stated, monatomic gases.
The applicability of the law of Dulong and Petit to the determination of atomic weights has been frequently exemplified during the last sixty years; and a number of these constants have been rectified by its means—e.g., of thallium, uranium, glucinum, indium, etc. The anomalies presented by the cases of elements of low atomic weight—e.g., carbon, boron, silicon—have been further inquired into; and it has been shown by Weber, and independently by Dewar, that in the case of these substances the specific heat rapidly increases with the temperature, and approximates at high temperatures to a value required by the law of Dulong and Petit.
Within recent years other methods of ascertaining molecular weights have been put at the disposal of chemists. These methods are especially valuable in the case of bodies which cannot be volatilised. They depend upon the influence of the substance (1) upon the freezing-point and (2) upon the boiling-point of a solvent. It has long been known that a substance in solution affects the freezing-point of the solvent, and in the great majority of cases depresses it.72 Sir Charles Blagden, as far back as 1788, showed that in aqueous solutions of inorganic salts the depression was proportional to the amount dissolved. It was subsequently found by Coppet that, in a number of solutions of similar salts where these were present in the ratio of their molecular weights, the solutions froze at practically the same temperature: the molecular depressions of the freezing-points differ from group to group but are nearly equal in groups of similar compounds. Raoult further observed that, when certain quantities of the same substance are successively dissolved in a solvent on which it exerts no chemical action, there is a progressive lowering of the point of solidification of the solution, and this depression is proportional to the weight of the substance dissolved in a constant weight of the solvent. In the case of a large number of solvents the depressions of the freezing-point, calculated for amounts proportional to the molecular weights of the dissolved substance, were nearly constant. Raoult pointed out that these relations between the molecular weights and the lowering in the freezing-point may be employed to determine the molecular weight of a soluble substance. The molecular weight m is found from the expression m = K/A, where A is the quotient obtained by dividing the observed depression in the freezing-point of the solvent by the percentage content of the solution,73 and K (the molecular depression) is a constant dependent on the solvent. Thus in the case of phosphorous oxide it was found that 0.6760 gram added to 20.698 grams of benzene—in which the oxide is soluble without change—lowered the freezing-point of the 3.16 per cent. benzene solution by 0°.68. Since the value of K for benzene is 49, we have (3.16 × 49)/0.68 = 227, which serves to indicate that P4O6 is the true molecular formula for phosphorous oxide. This result is confirmed by vapour-density observations.
The effect of adding a substance to a solvent is to diminish the vapour pressure of the liquid. Hence, since the boiling-point of a liquid is that temperature at which the vapour pressure is equal to the atmospheric pressure, the effect of adding the soluble substance is to raise the boiling-point, since a higher temperature is required in order that the pressure of the vapour shall equal that of the atmosphere. It has been proved that equal volumes of solutions in the same solvent which have the same boiling-point contain an equal number of molecules of the dissolved substance.
The equation for the molecular increment of the boiling-point for a solvent is d = 0.02T2/w, in which d is the increment of the boiling-point caused by the solution of one gram-molecule of a substance in 100 grams of the solvent, T the74 absolute boiling-point of the solvent, and w the heat of vaporisation of the solvent for one gram. The molecular rise of the boiling-point is therefore independent of the nature of the dissolved substance.
The molecular weight of the substance m is obtained from the formula m = pd/Δ, in which p = the percentage weight of the dissolved substance, d = the molecular increment in boiling-point (0.02T2/w), Δ = the observed rise in boiling-point. If the latent heat of vaporisation of the liquid is unknown, the value of d may be obtained by preliminary experiments with a substance of known molecular weight; in this case d = mΔ/p.
The calculation of the molecular weight m may also be made by the formula m = K(s/ΔL) × 100, in which Δ is the rise in boiling-point, s the weight of dissolved substance, L the weight of solvent, and K the molecular boiling-point increment. Convenient forms of apparatus for using these methods have been devised by Beckmann, and are now in general use.
From the time of Berzelius, each successive generation of chemists has striven to better the example of that master of determinative chemistry in the effort to obtain accurate values for the atomic weights of the elements.
75 Among the immediate successors of Berzelius in this work should be mentioned Turner, Penny, Dumas, and Marignac. Dumas in 1859 published the results of an extensive revision of the atomic weights of the elements. On this he based the far-reaching generalisation that, in the language of Prout, “the combining or atomic weights of bodies bear certain simple relations to one another, frequently by multiple, and consequently that many of them must necessarily be multiples of some one unit.” Dumas further agreed with Prout that “there seems to be no reason why bodies still lower in the scale than hydrogen (similarly, however, related to one another, as well as to those above hydrogen) may not exist, of which other bodies may be multiples, without being actually multiples of the intermediate hydrogen.”
The Belgian chemist, Stas, who had been associated with Dumas in a classical determination of the atomic weight of carbon, set himself to determine, with the highest degree of precision then possible, the atomic weights of about a dozen of the elements, with a view of ascertaining (1) whether an atomic weight is a definite and constant quantity, or whether, as suggested by Marignac, and subsequently by Crookes, an atomic weight represents “a mean value around which the actual weights of the atoms vary within certain narrow limits”;76 (2) whether, if the atomic weights of the elements are respectively definite and invariable, the numbers are commensurable as alleged by Prout and Dumas; and (3) if it should turn out that the numbers are severally fixed and commensurable, whether this necessarily indicates that the elements are built up of a primordial matter, the πρώτη ιλη of the ancients, referred to by Prout in 1816.
Stas devoted many years to the solution of these questions, working on a scale and with an accuracy and manipulative skill previously unapproached. The main results of his labour appeared in 1865. He concluded (1) that the atomic weights of the elements are absolutely constant values, and are not affected by the nature of the compounds in which they occur, or the physical conditions of their existence; (2) that the numbers so obtained are not commensurable: to quote his own words: “On doit considérer la loi de Prout comme une pure illusion.” Hence the elements must, on the basis of Stas’s experimental evidence, be regarded as “individualités à part,” as he expressed it—each a primordial and unalterable substance.
The appearance of this monumental work, which will ever remain one of the classics of chemistry, created a great impression. Its effect persists to this day. It constituted a77 model and furnished a standard which each successive worker has striven to emulate, with the result that atomic weights to-day are among the best ascertained of physical constants.
Space will not permit of any detailed account of the work done in connection with atomic weights during the forty-five years which have elapsed since the publication of Stas’s memoirs; and the reader who desires fuller information must be referred to the special treatises on the subject, such as the Constants of Nature of F. W. Clarke, or the monographs of Meyer and Seubert, Becker, Sebelien, and Van der Plaats.
Reference, however, must be made to the determinations by Lord Rayleigh, Leduc, Morley, Noyes, Guye, Dixon, and Edgar of values which, like those of oxygen, hydrogen, nitrogen, silver, and the halogens, are largely made use of as fiduciary values in atomic-weight work.
Lastly, it should be mentioned, the re-determination of the atomic weights of the elements with the highest attainable precision and by the most refined and most modern methods has for some years past been a special feature of the work of the Harvard Laboratory, under the direction of Theodore Richards; and some of the most trustworthy and best established values we possess have been ascertained by him and his pupils. Atomic weights are of such fundamental importance that the various nations78 interested in the pursuit of chemistry have consented to the establishment of an International Committee, which will take cognisance of the work done from time to time in this department of operative chemistry, examine and assess its value, and draw up an annual report on the subject.
Despite the accumulated testimony of this work in relation to the validity of the law of the conservation of mass, the sufficiency of the evidence has now and again been impugned. This aspect of the matter has within recent years been directly investigated by Landolt; and as the result of a painstaking series of experiments, in which every recognised source of error was removed or allowed for, it would appear that there is absolutely no ground for the belief that there is any dissipation of mass in the course of, or as the result of, a chemical change.
79
The more obvious physical phenomena of gases were, of course, well known by the middle of the nineteenth century; and the so-called gaseous laws—the laws of Boyle, Dalton, and Gay Lussac—were universally accepted by chemists and physicists at that time as fundamental. That the first two laws were only approximations to truth in a mathematical sense was also well known; and the experimental labours of Regnault and Magnus had not only established limits within which they were inexact, but had, to some extent, indicated the cause of their departure from the ideal condition. The hypothesis of Avogadro, as already stated, was practically ignored at this period, or at least its value was unappreciated until the time of Gerhardt and Laurent, and more particularly Cannizzaro, who in 1858 pointed out its real meaning and made it the keystone in the edifice of modern chemistry.
One of the most significant achievements of80 the last half-century has been the demonstration that these gaseous laws are interdependent. Their further study, and in particular the study of their variation from exact mathematical expression, has led to a conception of the real nature of a gas which not only comprehends and knits together these laws, but affords a rational explanation of them. If the laws of Boyle and Dalton concerning the relations between pressure, temperature, and gaseous volume are, and must be from the very nature of the case, only approximations, it follows that the same is equally true of the laws of Gay Lussac and Avogadro, since these are dependent on the others. The definite experimental proof that gases do not actually combine in the precise ratios demanded by the law of Gay Lussac has been forthcoming only during the last twenty years. It has been found that, instead of oxygen and hydrogen combining in the exact ratio of one volume of oxygen to two volumes of hydrogen to form water, as stated by Gay Lussac, one volume of oxygen combines with, according to Scott, 2.00245 vols.; according to Leduc, 2.0024 vols.; according to Morley, 2.00268 vols. of hydrogen. What is true of the volume-ratios in which oxygen and hydrogen are actually found to combine, under ordinary conditions, is no doubt equally true of analogous instances, such as the union of hydrogen and chlorine to form hydrogen81 chloride. It follows also that the extent of the variation from the mathematical expression of Gay Lussac’s law must get smaller and smaller as the combining gases approach the condition of the ideal gas—as they do, for example, under very low pressures. The precise degree of departure from Gay Lussac’s law is therefore in a sense accidental, and is dependent upon the conditions under which combination takes place.
The more exact study of the physical phenomena of gases, and in particular the clearer recognition of the causes which determine the extent of their departure from the ideal gaseous laws, have afforded valuable assistance in ascertaining the atomic weights of certain elements independently of chemical considerations. The processes of physical measurement have been so refined within recent years that physical methods of arriving at molecular—and, inferentially, at atomic—weights, in the case of all elements and compounds which can be brought into a condition approaching that of the ideal gas, are to be preferred to the gravimetric methods of analysis or synthesis as affording the most probable values of the true atomic weights of the elements. The work of Lord Rayleigh, Leduc, and of Guye and his pupils on the densities of the gases has furnished us with a series of values for the atomic weights of a number of the elements which, in point of accuracy, are as superior to82 the values of Stas as the values of Stas were superior to those of his predecessors. Daniel Berthelot pointed out in 1898 that the true molecular weight of a gas can be deduced from its density and its observed variations from Boyle’s law under atmospheric pressure and at very low pressures. Incidentally, the study of gaseous phenomena has served to place the theory of atoms upon a far more stable foundation than it occupied half a century ago. How halting was the adhesion which even some of the most eminent chemists then gave to this theory was well exemplified by the remarkable lecture given before the Chemical Society of London in 1869, in which Williamson—one of the most sturdy champions of Dalton’s doctrine—set forth its true value.
That a gas may be looked upon as an association of particles—hard elastic spheres—moving backwards and forwards in right lines with great velocity, and possessing in the aggregate a very small proportion of the space through which they travel, was first conceived by Daniel Bernoulli in 1738. By means of this hypothesis he explained the direct proportionality between the density and pressure of a gas. If the gas consists of moving particles, and the pressure which it exerts on the sides of the containing vessel is due to the impacts of these particles, it is obvious that by halving the83 original volume of the containing space we halve the space through which the particles travel, and therefore double the number of their impacts in a given time; in other words, by compressing the gas to half its initial volume we double the pressure it exerts, which is nothing else than the law of Boyle. This conception of the nature of a gas is known as the kinetic theory of gases; it was further developed by Waterston in 1845, still more fully by Clausius in 1857, and was subsequently placed in its present position by Maxwell and Boltzmann.
That gases actually do move, and at rates depending on their specific nature, was rendered probable—apart from this explanation of Boyle’s law—by many phenomena observed by chemists and physicists in the eighteenth and early part of the nineteenth century. It was known from the observations of Leslie in 1804 that specifically light gases moved or diffused faster than heavy gases. Attempts to determine these rates were made by Schmidt in 1820, and by Graham in 1846, both of whom found that the rate of movement of a gas was independent of its chemical nature, and was determined solely by its mass: gases move at rates inversely proportional to the square roots of their densities. The following table given by Graham shows the experimentally ascertained relative rates for a number of gases compared with the rates demanded by the84 “law of gaseous diffusion.” Column one gives the name of the gas; column two, the observed rate of diffusion; and column three, the square root of the density of the gas (air = 1):
| Gas. | Time of diffusion. | √density. |
|---|---|---|
| Air | 1 | 1 |
| Hydrogen | 0.276 | 0.263 |
| Marsh gas | 0.753 | 0.745 |
| Ethylene | 0.987 | 0.985 |
| Nitrogen | 0.986 | 0.986 |
| Oxygen | 1.053 | 1.051 |
| Carbon dioxide | 1.203 | 1.237 |
Nitrogen and ethylene are, chemically, totally dissimilar gases, but they have the same density and hence the same rate of movement. As Graham showed, it is possible to separate more or less completely a mixture of gases, if the constituents are of different densities, by taking advantage of their different rates of movement. Such an atmolytic method was employed by Rayleigh and Ramsay to prove that atmospheric nitrogen contained argon.
The fact that all gaseous substances, however different their chemical nature, conform in the main to certain simple “laws” indicates the probability that their mechanical structure is similar and comparatively simple. The so-called gaseous “laws”—the laws of Boyle,85 Dalton, Gay Lussac, Avogadro, and Graham—are to-day explained on the assumption that a gas consists of an aggregation of molecules, moving incessantly in straight lines and with great rapidity. The rate of movement of the particles is variable by reason of their mutual encounters; at the same instant some are moving rapidly, others more slowly. As already explained, to this ceaseless movement of the molecules is to be ascribed the pressure they exert; the pressure which a gas exerts on any containing surface is the aggregate effect of the impact of its molecules. The law of Boyle states that the product of the volume V and pressure P of a given mass of gas is invariable so long as the temperature is unchanged: PV = constant. It was found by Regnault, Magnus, Natterer, and Amagat that all gases, with the exception of hydrogen, show a departure from Boyle’s law in the sense that PV is less than theory demands. In the case of hydrogen PV is greater than theory. This exception, however, is only apparent. Every gas, if maintained above a certain temperature, shows, after a certain pressure has been reached, a deviation in the same sense as that exhibited by hydrogen.
The deviations from Boyle’s law are probably due to two causes: (1) to the effect of cohesion among the molecules, whereby the volume, and hence PV, is less than theory requires; (2) the86 molecules are not mathematical points—they have a certain volume; hence, with increasing pressure, PV is greater than theory demands. The effect of the molecule having a certain magnitude will be clear from the following figure: Let M be a molecule moving backwards and forwards within a certain space, a b:
Assume, now, we halve the containing space:
It will be seen that M, since its volume is unchanged, will have less than half the original distance to travel or, in other words, it will strike the boundaries of the containing space more than twice as frequently in the same interval of time as before; hence P, and therefore PV, becomes greater than Boyle’s law demands.
It will be noticed, then, that the two causes tending to bring about deviations from Boyle’s law act in contrary directions. In the greater number of gases the effect due to cohesion at ordinary pressure is greater than the effect due to the actual space occupied by the molecules. In the case of hydrogen at ordinary temperature the contrary is the case; if, however, hydrogen is strongly cooled, it shows variations similar to those exhibited by other gases at ordinary temperatures.87 By heating these gases the effect due to cohesion—to the mutual attraction of the molecules—becomes less and less; in such circumstances these gases show departures from theory in the same sense that hydrogen does at ordinary temperatures.
The effect of mutual attraction among the molecules is to make the volume of the gas less than the theoretical value; the cohesive force may therefore be regarded as equal in effect to a certain additional pressure; that is, (P + A) V = constant, in which A is the measure of the force of cohesion. A, of course, must have relation to the number of molecules mutually attracted: A is proportional to the square of the number of the molecules. But the number of the molecules in the unit volume is proportional to the density of the gas, and in a given mass of gas the density is inversely proportional to the volume. Hence A is inversely proportional to the square of the volume—A = a/V2, hence (P + a/V2) V = constant. Now let us trace the effect of the second cause of variation from the mathematical exactitude of Boyle’s law. The fact that the molecules are not mathematical points means that V in the foregoing expression is not identical with the space in which the molecules move. That space is V - b, in which b is the measure of the aggregate volume of the molecules. Hence the true 88expression becomes (P + a/V2) (V - b) = constant.
The law of Dalton (Charles) also receives its simplest explanation by the kinetic theory of gases; and, moreover, the departures from the mathematical truth of the statement follow as a necessary consequence of the facts that the molecules have sensible magnitudes and are mutually attracted. We can measure the effect of heat upon a gas in two ways. We can either keep the pressure of the gas constant, and measure the increase in volume; or we can prevent the gas from expanding, and measure the elastic force or pressure it exerts. If the law of Dalton were mathematically true, it would follow that, if the volume of the gas were maintained constant during the heating, its pressure would increase in the same proportion as the volume would have increased if the gas had been allowed to expand, but maintained at a constant pressure. In other words, the expansion-coefficient and the pressure-coefficient should be the same. Experiment shows, however, that they are not identical.
The following table gives the results of a number of measurements by Regnault:
| Expansion (Pressure Constant). | Pressure (Volume Constant). | |
|---|---|---|
| Hydrogen | .003661 | .003667 |
| Air | .003670 | .003665 |
| Carbon dioxide | .003710 | .003688 |
| Sulphur dioxide | .003903 | .003845 |
89 Variations in the same sense have since been observed by Jolly and Chappuis. With the exception of hydrogen, and probably also helium, all the gases show greater values for the coefficient of expansion than for the coefficient of pressure, and the differences are greater the greater the coefficient of expansion of the gas.
Since, as we have already stated, the law of Boyle is directly related to the law of Dalton, both being dependent on molecular movements, the same course of reasoning used to account for the variations in the case of Boyle’s law applies equally to the case of Dalton’s law. The “law” of Avogadro follows also as a necessary consequence of this explanation of the laws of gaseous pressure and temperature. If all gases show approximately the same increase in pressure when heated under constant volume, and if the increased pressure is due only to the increased energy with which the molecules strike the sides of the containing vessel, it follows that all gases must contain the same number of molecules in unit volume. But as, from the very nature of the case, the laws of Boyle and Dalton cannot be mathematically true, it follows that the laws of Avogadro and Gay Lussac must be only approximations in the same sense.
The law of Graham, connecting the rate of diffusion of a gas with its density, follows also as a necessary consequence of this explanation90 of the laws of Boyle, Dalton, Gay Lussac, and Avogadro. If the number of molecules in the unit volume of any gas, whatever be its nature and whatever be their mass, is approximately the same, it follows that the mean velocity of the molecules must be variable; their mean velocities must be in the inverse ratio of the square roots of their densities.
The mean velocity with which the molecules of a gas move can be calculated if we know the pressure it exerts, the weight of a definite volume, and the value of the acceleration due to gravity. The square of this velocity in metres per second of time at 0°C. is given by the expression U2 = 3pg/q in which p = pressure per square metre = 10,333 kilograms; g = the gravitation constant = 9.81; q = weight of a cubic metre of the gas at 0°C. and one atmosphere of pressure.
For hydrogen we have
U2 = 3 × 10,333 × 9.81/0.0899
whence U = 1842 metres per second; for oxygen we have U2 = 3 × 10,333 × 9.81/1.430, whence U = 461. These numbers accord with those demanded by Graham’s law. The density of H being taken as 1, that of oxygen is 16 and √16 = 4; the numbers 1842 and 461 are in the ratio of 4 to 1.
The amount of heat required to raise the temperature of the unit mass of a gas through91 a definite interval depends, as Laplace first pointed out, upon whether the gas is allowed to expand or not; in other words, the specific heat of a gas varies as the heating is at constant volume or at constant pressure. If, having raised the temperature of the unit mass, and so expanded it, we then compress it until it occupies its initial volume, a further rise of temperature takes place without any external heat having been applied. This rise of temperature is, in fact, due to the liberation of the amount of heat required merely to expand the gas without increasing its temperature. The quantity of heat needed to raise the temperature of a gas through a definite interval is therefore greater when it is allowed to expand than when its volume is kept constant; in other words, the specific heat at constant pressure is greater than the specific heat at constant volume. The ratio of the two specific heats can be calculated: on the assumption that the energy imparted to the molecules simply accelerates their mean rectilinear velocity, and that no energy is absorbed in doing internal work among them, it is found that, when the gas is permitted to expand, the amount of heat required is 1.67 times greater than that needed when its volume is kept constant. This ratio has been experimentally determined for a number of gases. For oxygen under normal conditions it is 1.408, for hydrogen 1.414, for carbon dioxide 1.264,92 for methane 1.269—all numbers notably below the value 1.67. The direct experimental determination of this ratio by thermometric measurements is a matter of some difficulty. It was, however, demonstrated by Dulong that it can be ascertained with comparative ease from observations on the velocity of sound in the gas—the velocity being probably a direct function of this ratio. As carried out experimentally, the method consists in sending a sound-wave through the gas contained in a glass tube along the horizontal length of which is strewn a quantity of a light powder such as the spores of lycopodium or finely divided silica. The glass tube is fitted at one end with a glass rod; by rubbing this a series of longitudinal vibrations is set up and communicated to the gas whereby the light powder is thrown up into little heaps along the tube, the distance between the heaps being equal to half a wave length. By comparative measurements with air and the gas under examination, data are obtained from which the ratio of the specific heats can be deduced.
By experiments conducted on this principle Kundt and Warburg found that mercury vapour gave numbers agreeing with the theoretical ratio 1.67. Now, its vapour density shows that mercury vapour is a monatomic gas; it actually fulfils the conditions prescribed for a gas which93 theory indicates should give the value 1.67. All the energy imparted to its molecules on heating simply accelerates their translational velocity. On the other hand, all the gases above named as giving values below 1.67 are diatomic gases; in their case the energy imparted to them is employed partly in augmenting the translational velocity of the molecules, and partly in bringing about internal changes within them. By experiments made in like manner Ramsay and Travers succeeded in showing that the inert gases of the atmosphere are monatomic.
No attempt can be made here to explain the various methods by which it has been sought to obtain an estimate of the absolute size of gaseous molecules or to determine their number in a definite volume. By observations on their viscosity, rates of diffusion, conductivity for heat, variations from the law of Boyle, dielectric constants, electric charges, etc., Maxwell, O. E. Meyer, Loschmidt, Lothar Meyer, Van der Waals, Mossotti, Planck, Sir J. J. Thomson, and others, have arrived at estimates of the magnitude and number of molecules in a gas. These estimates necessarily vary with the hypotheses made in deducing them. It would serve no useful purpose to give the results, since the figures convey no impression to the mind of the minuteness of molecules, or even as to the extraordinary number of them in, say, so small94 a volume as one cubic centimetre. As an example, it has been calculated that there are about 640 trillions of hydrogen molecules in one milligram of the gas.3
3 O. E. Meyer, The Kinetic Theory of Gases, 1899.
In the preceding volume a short account has been given of the history of the early attempts to effect the liquefaction of the gases. These resulted in their division into the two classes of liquefiable and permanent gases. One of the most notable achievements of the latter half of the last century was to sweep away this arbitrary distinction. The fundamental condition needed to effect the liquefaction of a gas, although surmised by Faraday, was first clearly indicated by Andrews about 1863. He showed that, in order to liquefy a gas, its temperature must be lowered to a point peculiar to each gas, when, on the application of sufficient pressure, it will become a liquid. Thus, in the case of gaseous carbon dioxide, Andrews found that, if its temperature were maintained above 31° C., no amount of pressure would cause it to liquefy; if the temperature were lowered just below this point—termed the critical point—a pressure of 75 atmospheres would effect its liquefaction. On the other hand, if the temperature of the liquid carbon dioxide be slowly raised to about 31°, the surface of demarcation95 between the liquid and the gas becomes gradually fainter and eventually disappears. Carbon dioxide may thus be made to pass from the state of liquid to that of gas without any sudden alteration of volume. If a given volume of the gas, say at 50°, be exposed to gradually increasing pressure, say up to 150 atmospheres, the volume is gradually diminished with the increment of pressure, but no sudden contraction indicating liquefaction occurs. If the gas under the high pressure be allowed to cool down to the ordinary temperature, no sudden contraction is observed to follow. The carbon dioxide, at the outset a gas, in the end becomes a liquid by a gradual and continuous transition, unaccompanied by any abrupt change of volume. These observations show that what we style the liquid and gaseous states are simply separated manifestations of the same condition of matter. There is a definite temperature for every gaseous substance at which it ceases to be liquefiable under pressure; and the reason that Faraday failed to liquefy certain gases was that he was unable, with the means at his command, to lower their temperatures sufficiently and so reach their critical points; hence the enormous pressures which he and other investigators applied were unavailing. These facts were definitely made known by Andrews in 1869, were theoretically developed by Van der Waals96 in 1873, and practically applied to the liquefaction of oxygen in 1877, independently and almost simultaneously, by Pictet, of Geneva, and Cailletet, of Châtillon-sur-Seine. Pictet exposed oxygen, under great pressure, to the cold produced by the rapid evaporation of liquid carbon dioxide; Cailletet brought about the same result by suddenly diminishing the tension of the strongly compressed oxygen, the rapid expansion of the gas effecting the reduction of its temperature below the critical point. Other workers took up the subject, notably Wroblewski and Olszewki in Poland, Dewar in England, and Kammerlingh Onnes in Holland; and the liquefaction of all the gases has now been accomplished.
The following table shows the absolute boiling-(B.P.) and melting-points (M.P.), critical points (C.P.), and pressures (C.Press.), together with the density (D) at their boiling-points of a number of liquefied gases:
| B. P. degrees | M. P. degrees | C. P. degrees | C. Press. m. | D. | |
|---|---|---|---|---|---|
| Helium | 4.5 | — | — | — | 0.15 |
| Hydrogen | 20 | 15 | 35 | 11.6 | 0.06 |
| Oxygen | 90.5 | below 50 | 154 | 44 | 1.131 |
| Nitrogen | 77.5 | 60 | 124 | 20.9 | 0.791 |
| Methane | 108.3 | — | 191 | 42.4 | 0.416 |
| Ethylene | 169.5 | 104 | 282 | 44 | 0.571 |
| Fluorine | 186 | 40 | — | — | 1.11 97 |
| Chlorine | 239.6 | — | — | — | 1.507 |
| Ammonia | 234.5 | 197.5 | 404 | 85.9 | — |
| Neon | 30.40 | — | below 65 | — | — |
| Argon | 86.90 | — | 155.6 | 40.2 | 1.212 |
| Krypton | 121.33 | — | 210.5 | 41.2 | 2.155 |
| Xenon | 163.9 | — | 287.8 | 43.5 | 3.52 |
The principle of the Cailletet method of effecting the liquefaction of oxygen had been theoretically and experimentally studied by Joule and Lord Kelvin many years previously. It was extended by Siemens and has been applied by Linde and Hampson to the construction of machinery for the production of liquid air on a large scale, without the use of any intermediate refrigerant.
It is now readily possible to procure considerable quantities of liquid air, and even of liquid hydrogen. By the evaporation of liquid hydrogen temperatures approaching the absolute zero—that is, 273° C. below the melting-point of ice—can now be reached. Incidentally there has been developed a special field of inquiry relating to the behaviour of substances at low temperatures.

The pioneers in this field have been Dewar in England and Kammerlingh Onnes in Holland.9899 Research at low temperatures, indeed, has been the main feature of the work of the Royal Institution of Great Britain during the last twenty years. It has included observations at temperatures approaching the absolute zero, on the electrical resistivity of metals and alloys, on the behaviour of so-called insulators, on changes in the cohesive force of metals, on the dielectric constants of frozen electrolytes, on the influence of cold on magnetisation and on magnetic permeability, and on the optical behaviour of bodies, on vital phenomena at low temperatures, and on the influence of cold on chemical change.
Dewar has succeeded in liquefying and solidifying large quantities of hydrogen, and has studied its properties at low temperatures. Liquid hydrogen is transparent and colourless. It is a non-conductor of electricity, and gives no absorption spectrum. It freezes into an ice-like solid, devoid of metallic properties. Dewar has made use of the property possessed by charcoal of occluding gases, especially at low temperatures, in the production of high vacua, and in the separation of gases; and he has also determined the molecular heat of absorption by charcoal of various gases. He has employed liquid air, liquid nitrogen, and liquid hydrogen as calorimetric agents, and has determined by means of them the heat capacities of a number of100 substances at very low temperatures. Lastly, his ingenious contrivance of silvered vacuum protected vessels, now introduced into commerce under the name of “Thermos flasks,” has greatly facilitated the manipulation of liquefied gases for experimental purposes.
101
In an anonymous essay “On the Relation between the Specific Gravities of Bodies in their Gaseous State and the Weights of their Atoms,” published in Thomson’s Annals of Philosophy in 1815, the attempt was made to indicate certain consequences which seem to follow from Dalton’s law of gaseous volumes, as generalised by Gay Lussac. The author of this essay was subsequently discovered to be a medical student named William Prout, noteworthy as having been one of the first to point out the suggestiveness of the numerical relationships which occur among the atomic weights of the elements. This paper is usually assumed to contain the statement that the atomic weights of the elements are multiples of that of hydrogen. As a fact, however, this hypothesis is nowhere explicitly stated in the paper. The inference was practically due to Thomson, who strove to support it by experimental proof of so weak a character as to draw forth the remark of Berzelius102 that much of it appeared to have been made at the writing-desk.
Nevertheless, the occurrence of such numerical relationships continued, as already stated, to excite speculation. Döbereiner, in 1829, pointed out that in certain groups of correlated elements, consisting each of three members, the middle member had an atomic weight practically identical with the arithmetic mean of the atomic weights of the others; and similar observations were made by Gmelin, Dumas, Gladstone, and Strecker. An approach to the recognition of the general law underlying these facts was made by Newlands in England, and independently by De Chancourtois in France, who were the first to indicate that the properties of the elements are related to their atomic weights. This conception was developed by the Russian chemist, Mendeléeff. In Mendeléeff’s arrangement, first published in 1869, the elements are so grouped that their properties are periodic functions of their atomic weights. The general statement of what is now known as the Periodic Law may be put in this form: If the elements are arranged in order of increasing atomic weight, the properties of these elements vary from member to member of the series, but return more or less nearly to the same value at certain fixed points in the series. This is observed to occur in the atomic value, or valency, of the103 several members; also in their specific volumes, melting-points, ductility, hardness, volatility, crystalline form, thermal expansion, refraction equivalents, and conductivities for heat and electricity, in their magnetic properties and electro-chemical behaviour, and in their heats of chemical combination, etc.
The first chemist of note to grasp the significance of Mendeléeff’s generalisation was Lothar Meyer, who, dealing at the outset with one of the characteristic properties of the elements—viz., their specific or atomic volumes (that is, the values obtained by dividing their specific gravities into their respective atomic weights)—greatly developed the principle of periodicity, representing it graphically in a most striking and suggestive manner, leading up to a classification almost identical with that of Mendeléeff.
Since the date of its promulgation the scheme of classification of the elements in accordance with the principle of periodicity has experienced certain minor modifications necessitated by fuller knowledge; but in its essential features it remains very much in the form devised by Mendeléeff. The discovery of the so-called inert and radio-active elements required that their relations to the periodic law should be defined. Their inclusion raises no fundamental difficulty. Indeed, the generalisation seems to adapt itself to the far-reaching considerations which spring104 from modern views of the nature of the atom, its electro-chemical relationships, and the orderly arrangement of the corpuscles of which it may be composed. In the 1905 edition of the English translation of his famous Principles of Chemistry, Mendeléeff has given a table which may be said to embody his final views concerning the systematic classification of the elements. This is reproduced on p. 105. In this table he postulates the existence of two hypothetical elements, x and y, the former of which he regards as identical with the physical ether; while the latter is an analogue of helium, possibly identical with the “coronium” of the solar coronal atmosphere, with a molecular weight of about 0.4.
The striking feature of Mendeléeff’s generalisation is its universality. In this respect it differs from all previous attempts at natural classifications of the elements; these were limited and partial, and therefore unsatisfactory. Nevertheless, it is easy to trace in them fundamental conceptions upon which Mendeléeff built. Mendeléeff, in fact, gave a great extension to ideas with which the chemical world of half a century ago was more or less familiar; and doubtless it was this circumstance, combined with the remarkable boldness and comprehensiveness of this extension, followed almost immediately by a most striking series of confirmations of his own previsions, as logical105 consequences of his generalisation, that secured for it attention, and ultimately universal adoption.
| Series. | Zero group. | Group I. | Group II. | Group III. | Group IV. | Group V. | Group VI. | Group VII. | Group VIII. | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | x | — | — | — | — | — | — | — | — | |
| 1 | y | Hydrogen. H = 1.008 |
— | — | — | — | — | — | ||
| 2 | Helium. He = 4.0 |
Lithium. Li = 7.03 |
Beryllium. Be = 9.1 |
Boron. B = 11.0 |
Carbon. C = 12.0 |
Nitrogen. N = 14.04 |
Oxygen. O = 16.0 |
Fluorine. F = 19.0 |
— | |
| 3 | Neon. Ne = 19.9 |
Sodium. a = 23.05 |
Magnesium. Mg = 24.1 |
Aluminium. Al = 27.0 |
Silicon. Si = 28.4 |
Phosphorus. P = 31.0 |
Sulphur. S = 32.06 |
Chlorine. Cl = 35.45 |
— | |
| 4 | Argon. Ar = 38 |
Potassium. K = 39.1 |
Calcium. Ca = 40.1 |
Scandium. Sc = 44.1 |
Titanium. Ti = 48.1 |
Vanadium. V = 51.4 |
Chromium. Cr = 52.1 |
Manganese. Mn = 55.0 |
{ | Iron. Fe = 55.9 Cobalt. Co = 59 Nickel. Ni = 59(Cu) |
| 5 | — | Copper. Cu = 63.6 |
Zinc. Zn = 65.4 |
Gallium. Ga = 70.0 |
Germanium. Ge = 72.3 |
Arsenic. As = 75.0 |
Selenium. Se = 79.0 |
Bromine. Br = 79.95 |
— | |
| 6 | Krypton. Kr=81.8 |
Rubidium. Rb = 85.4 |
Strontium. Sr = 87.6 |
Yttrium. Y = 89.0 |
Zirconium. Zr = 90.6 |
Niobium. Nb = 94.0 |
Molybdenum. Mo = 96.0 |
— | { | Ruthenium. Ru = 101.7 Rhodium. Rh = 103.0 Palladium. Pd = 106.5(Ag) |
| 7 | — | Silver. Ag = 107.9 |
Cadmium. Cd = 112.4 |
Indium. In = 114.0 |
Tin. Sn = 119.0 |
Antimony. Sb = 120.0 |
Tellurium. Te = 127 |
Iodine. I = 127 |
— | |
| 8 | Xenon. Xe = 128 |
Cæsium. Cs = 132.2 |
Barium. Ba = 137.4 |
Lanthanum. La = 139 |
Cerium. Ce = 140 |
— | — | — | — | |
| 9 | — | — | — | — | — | — | — | — | — | |
| 10 | — | — | — | Ytterbium. Yb = 173 |
— | Tantalum. Ta = 183.0 |
Tungsten. W = 184 |
— | { | Osmium. Os = 191 Iridium. Ir = 193 Platinum. Pt = 194.9(Au) |
| 11 | — | Gold. Au = 197.2 |
Mercury. Hg = 200.0 |
Thallium. Tl = 204.1 |
Lead. Pb = 206.9 |
Bismuth. Bi = 208 |
— | — | — | |
| 12 | — | — | Radium. Rd = 224 |
— | Thorium. Th = 232 |
— | Uranium. U = 239 |
— | — |
106 The periodic law, in the words of its author, is the “direct outcome of the stock of generalisations of established facts which had been accumulated by the end of the decade 1860–1870.” It is founded wholly on experiment, and is as much the embodiment of fact as are the laws of chemical combination. It was based upon the adoption of the definite numerical values of the atomic weights, as indicated by Cannizzaro, as a consequence of the hypothesis of Avogadro, and upon the assumption that the relations between the atomic weights of analogous elements must be governed by a general law. The application of the periodic law immediately led to the re-determination of certain atomic weights and to the correction of their assumed atomic values. At the time of its enunciation the determination of the valency of an element was purely empirical, with no apparent necessary relation to that of other elements. We find now that the valency is a matter of a priori knowledge, just as much as any other property of the element. The amended values for the atomic weight and valency of a number of elements thus demanded by the law have been confirmed by all the experimental criteria employed by chemists. The generalisation further indicated107 the existence of new elements; it pointed out their probable sources, and foretold their properties. Instances of this power of divination in the law are to be seen, as already mentioned, in the discovery of gallium by Lecoq de Boisbaudran, of scandium by Nilson, and of germanium by Winkler, the existence and main properties of which were severally foretold by Mendeléeff in 1871.
The promulgation of the law was heralded as a proof of the validity of the conception of a primordial matter. It was held that it can find a rational explanation only in the idea of unity in the formative material. But its author would not admit that his generalisation had any relation to the Pythagorean hypothesis:
The periodic law, based as it is on the solid and wholesome ground of experimental research, has been evolved independently of any conception as to the nature of the elements. It does not in the least originate in the idea of an unique matter, and it has no historical connection with that relic of the torments of classical thought; and therefore it affords no more indication of the unity of matter or of the compound nature of the elements than do the laws of Avogadro, and Gerhardt, or the law of specific heats, or even the conclusions of spectrum analysis. None of the advocates of a unique matter has ever tried to explain the law from the standpoint of ideas taken from a remote108 antiquity, when it was found convenient to admit the existence of many gods—and of a unique matter.
The reader who desires a fuller exposition of the principles of the periodic law must be referred to special treatises on the subject, or to the larger manuals on general chemistry. It must, however, be stated that, while many facts discovered since the original promulgation of the principle and since its development by Lothar Meyer, Carnelley, Thomsen, and others, are consistent with the law, other facts, some of which were known before 1870, are apparently out of harmony with it, or at all events await a fuller interpretation. For example, tellurium is not in its proper place in the scheme if its atomic weight, 127.5, has been correctly ascertained. Cobalt (58.97) and nickel (58.68) have atomic weights so closely accordant that their properties and those of their corresponding compounds should be very similar, and, in fact, almost identical; but such is not the case. Indeed, it has been said, no prevision of the periodic law would have led to the discovery of nickel. Similar considerations apply to manganese, chromium, and iron; the atomic weights of these elements are less widely different than the differences in their properties and the divergence in their chemical relationships would seem to109 require. The relative positions of argon and potassium are also not consistent with the law. There are difficulties, too, connected with what we know at present concerning the atomic weights of the so-called rare earth metals. In spite, however, of these seeming anomalies, it can hardly be doubted that the periodic law is as much the expression of a natural law as is the law of gravitation; although it is possible, and indeed probable, that, as we now define it, it is only the first approximation to the truth, and that, as our knowledge becomes more precise, Mendeléeff’s classification, in its present form, will require modification and extension, just as Mendeléeff’s own scheme may be said to be a modification and extension of the attempts at the rational classification of the chemical elements made by his predecessors.

Dmitri Ivanowitsch Mendeléeff, with whose name this fruitful generalisation is indissolubly connected, was born February 7, 1834 (N.S.), at Tobolsk, in Siberia, and was the fourteenth and youngest child of Ivan Mendeléeff, the Director of the gymnasium at that place. Soon after the birth of Dmitri his father became blind, and the family were practically dependent upon the mother, Maria Dmitrievna Mendeleeva, who established a glass works near Tobolsk, on the profits of which she brought up and educated her large family. At the age of fifteen Mendeléeff110111 was taken by his mother to St. Petersburg, and began the study of natural science at the Physico-Mathematical Faculty of the Institute. After serving as a science master at Simferopol in the Crimea and at Odessa, in 1856 he became a privat-docent in the University; then, following a short period of study in France and Germany, he returned to St. Petersburg, and in 1866 he was made Professor of General Chemistry in the University. His reputation mainly rests upon his contributions to chemical philosophy and physical chemistry, notably on specific volumes, on critical temperatures, on the thermal expansion of liquids, on the nature of solutions, on the elasticity of gases, and the origin and nature of petroleum. He died on January 31, 1907.
112
Chemical formulæ, from the time of Berzelius onwards, have been regarded as rational expressions—that is, they serve to represent the relations and analogies of the substance they are employed to designate, and indicate in the simplest and at the same time the most comprehensive manner the chemical changes in which the substances take part. In the words of Gerhardt, those formulæ are “the best that make evident the greatest number of such relations and analogies,” and that serve to express the greatest number of the chemical changes in which they are concerned.
In such concrete expressions of chemical change it was frequently observed that a definite group of some or all of the constituent elements of the substance hung together, as it were, and passed, apparently unchanged, into the products of its transformation. These groups were not necessarily radicals in the sense in which Liebig and Wöhler used the term; to113 Gerhardt and to Kekulé they were simply residues, remaining unattacked in a chemical metamorphosis, and passing as such into the products of the change. They might or might not be capable of isolation as definite entities. Thus, for example, we may represent the composition of the following sulphur compounds so as to show that they all contain the group SO2, or sulphuryl:
These formulæ serve to show how the several substances are mutually related, and that they may be derived from one another by the substitution of atoms of chlorine for hydroxyl (OH), or nitryl (NO2), or vice versa.
It was pointed out in 1851 by Williamson, and subsequently by Gerhardt, that these groups are characterised by differences in their power of combining with or replacing atoms of hydrogen, or of groups or elements which, like chlorine, are chemically equivalent to hydrogen. Such 114a radical or residue as ethyl (C2H5) is chemically equivalent to one atom of hydrogen, as is shown when we compare the formula for ether, as established by Williamson, with that of ordinary alcohol:
Sulphuryl, SO2, is chemically equivalento two atoms of hydrogen; phosphoryl, PO, as suggested by Odling, to three atoms of hydrogen. Gerhardt therefore proposed to designate these and similar groups as monatomic, diatomic, triatomic, according to their respective hydrogen-replacing power.
This conception of the definite atom-fixing or replacing power of groups or compound radicals was extended by Frankland, in 1852, so as to include the simple radical—that is, the elements. In the memoir in which he announced the existence of the organometallic compounds he pointed out that the elements may be classified according to their combining power, or, as he expressed it, according as “their affinities are best satisfied.” This idea was independently developed by Couper and Kekulé in 1858; it is from that period that the definite introduction of the conception of atomicity, atomic-value, or valency, into chemical doctrine may be said to date.
115 The memoir in Liebig’s Annalen der Chemie und Pharmacie, in which Kekulé announced his views, deals particularly with the tetravalency of carbon and the doctrine of linking of atoms in terms of their valency. As formulated by Kekulé and as subsequently developed in his famous text-book, this doctrine exercised an immediate effect on the progress of the chemistry of carbon compounds. Like every fruitful hypothesis, it stimulated inquiry, and brought out analogies; and the more it was applied the more apparent became its suggestiveness and utility. The scope of chemical formulæ was greatly extended. Rational formulæ grew into dissected or constitutional formulæ; and on the system of constitutional formulæ have been grafted successive attempts to elucidate the manner in which the constituents of a molecule are grouped and held together. It is interesting to note that the proximate effect of the theory of chemical structure which grew out of Kekulé’s doctrine was to assimilate what was sound in the seemingly antagonistic theories of types and radicals. As a mode of exposition, Kekulé used models to illustrate the manner in which the affinity-values of compounds are satisfied; these were not intended to represent the actual spatial distribution of the atoms in a molecule, but they nevertheless familiarised the mind with the idea first clearly recognised by Wollaston116 and Berzelius that this is the ultimate aim of chemistry. It was probably their use, either actually or by visualisation, that led Kekulé in 1865 to his theory of the constitution of benzene, as developed in his paper on the constitution of the aromatic compounds—a theory no less fruitful in its consequences than that of the tetravalency of carbon and of the linkage of atoms. Such models, too, in the hands of Van ’t Hoff, subsequently served to elucidate the connection between optical characters and crystalline form, and to explain the isomerism of certain organic substances.
Kekulé was of opinion that the valency, or affinity-value, of an element was a definite and invariable quantity—a fundamental property of the atom as immutable as its atomic weight. Many facts appear to show that such is not the case. Thus phosphorus and nitrogen are sometimes trivalent and at other times pentavalent; tin, in certain of its compounds, is divalent; in others, tetravalent. Sulphur may be a dyad, a tetrad, or a hexad. It will be seen that the valency of these particular elements varies by two units: this was at one time held to be a natural law, and the various elements were divided by Frankland into the two main groups of (1) perissads, or elements of odd atomic value, and (2) artiads, or elements of even atomic value. Experience has demonstrated that a117 rigid classification on this basis is not possible. Many instances are known of elements not only varying in valency by two units, but even by one unit. Thus nitrogen, which is usually a perissad, is apparently an artiad in nitric oxide and in gaseous nitrogen peroxide. Roscoe has shown that uranium and tungsten, originally regarded as artiads, form pentachlorides.
To what the difference in the affinity-value of an element is due, and why different elements should manifest different values, is at present unknown. Valency, like other properties, appears to be a periodic function of atomic weight; from the behaviour of such analogous compounds as phosphorus pentafluoride, phosphorus pentachloride, phosphorus pentabromide, it seems, too, to be related to the weights of the atoms in combination. Further, it would appear that the mutual affinities of substances vary with temperature—i.e., with the energy imparted to their molecules; numerous instances appear to indicate that the atom-fixing power of an element decreases when it is strongly heated—that is, when the internal energy imparted to its combinations exceeds a certain limiting value. Van ’t Hoff has attempted a mechanical explanation of valency depending on the shape of the atoms, as affected by variation in their vibratory motions resulting from differences of temperature. Helmholtz suggested that the118 different charges of electricity associated with the atoms may determine their affinity-values—that, for example, a monad carries a single charge, a dyad two, a triad three charges. Many considerations go to show that the affinity-value of an element is not capable of definite numerical expression in the sense which the doctrine of valency as generally understood implies, and that the variations are not of the per saltum character assumed by saying that the affinity-value is sometimes 1, sometimes 2, at other times 3, and so on. When we have apparently satisfied the accepted atomic value of an element by allocating to it what we regard as the necessary complement of atoms of other bodies, it is frequently evident that the capacity for combination of the whole molecule is not satisfied. Many apparently saturated molecules have the power of combining with other equally saturated molecules. Sulphur trioxide (SO3) and barium monoxide (BaO) would appear each to have their affinity-values satisfied; nevertheless they combine with great 119readiness to form barium sulphate, BaSO4.
The suggestions of Couper and Kekulé that an explanation of the properties of chemical compounds should be sought in the nature and mutual affinities of their constituent elements rather than of their radicals were not wholly accepted at the time they were first made. Speculative ideas have to justify themselves by facts. The value of an hypothesis depends upon its usefulness and expediency, and on its power of indicating the lines of future inquiry. How far it is inductively sound and deductively useful is a matter for individual judgment. Consequently the tendency to pass from purely rational and constitutional formulæ to formulæ which sought to symbolise the inner structure—the very skeleton, as it were—of a molecule, was resisted for a time, and by no one more strongly than by Kolbe.
Kolbe’s attitude to the new doctrine may be said to have had its justification in his own work. His remarkable prediction, based on120 considerations which had nothing in common with Kekulé’s doctrine, of the existence of the secondary and tertiary alcohols, so soon to be confirmed by Friedel’s discovery of secondary propyl alcohol, and by Butlerow’s isolation of tertiary butyl alcohol, served to retard the general adoption of Kekulé’s views by showing that apparently they were no more fruitful in suggestiveness than those they were intended to supplant. But it was exactly in their suggestiveness with regard to the development of isomerism that structural formulæ based upon valency were gradually found to be most useful. It was perceived that it was now possible not only to foretell the existence of isomers, but to determine their number, and to some extent to forecast their properties and modes of decomposition. Cayley, for example, calculated the number of possible isomers of the hydrocarbons of the generic formula CnH2n+2 up to C6H14 all those that theory predicted have been discovered. In no single case have more been obtained than the number indicated by theory. The accumulated weight of this and similar testimony ultimately established the doctrine of chemical structure on a firm basis.
This conception received a great extension as the result of Kekulé’s application of his ideas to the explanation of the chemical constitution121 of the group of substances of vegetable origin—consisting of essential oils, balsams, resins, and their products, which, on account of their characteristic odours, were collectively known to the older chemists as the aromatic compounds. Some of these, like oil of bitter almonds, gum benzoin, coumarin, oil of wintergreen, oil of anise, oil of cinnamon, oil of cumin, balsam of tolu, phenol, and certain of their derivatives, such as benzene, aniline, salicylic acid, cinnamic acid, toluene, cymene, had already been investigated with important theoretical results; but as a class they had received far less attention than the derivatives of the great group of homologous radicals of which methyl is the initial member. Of course it was part of the doctrine of Liebig—the discoverer of benzoyl—that the aromatic compounds also contained specific radicals; but the relation of these compounds to those we now call aliphatic (fatty) compounds was not understood, although certain analogies were recognised.
In 1866 Kekulé drew attention to the following significant peculiarities of the aromatic compounds: (1) All aromatic compounds, even the simplest, are comparatively richer in carbon than the corresponding class of fatty (aliphatic) compounds; (2) among the aromatic substances, as among fatty compounds, numerous homologous compounds exist; (3) the simplest aromatic122 substances contain at least six atoms of carbon; (4) all decomposition products of aromatic substances show a certain family resemblance; the main product of the decomposition contains at least six atoms of carbon—e.g., benzene C6H6, phenol C6H6O, etc., which would seem to indicate that all aromatic substances contain a nucleus or atomic grouping containing six carbon atoms. Within this nucleus the carbon atoms are in closer connection or denser combination, from which it follows that all aromatic compounds are comparatively rich in carbon. More carbon atoms can then be added to this nucleus according to the same laws that govern the fatty compounds. In this way the existence of homologous compounds may be explained.
On the assumption that carbon is tetravalent and that its valency is constant, Kekulé showed how, by linking together six carbon atoms by alternate single and double bonds, six affinity units may be left free. If we assume that six carbon atoms are attached to one another according to this law of symmetry, we obtain a group which, regarded as an open chain, contains eight unsaturated units of affinity:
By making the further assumption that the two carbon atoms at the ends of the chain are linked123 together by one unit of affinity each, a closed chain (a symmetrical ring) is obtained which now contains six unsaturated units of affinity:
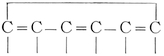
From this closed chain all the substances usually designated as “aromatic compounds” are derived. In these a common nucleus may be assumed: it is the closed chain C6A6, where A denotes an unsaturated affinity. The six affinities of this nucleus may be satisfied by six monovalent elements. They may also, wholly or in part, be satisfied by one affinity of polyvalent elements, the latter necessarily bringing with them other atoms into the compound, thus producing one or more side chains, which in their turn may be lengthened by the addition of other atoms.
If each of the free units is satisfied by an atom of hydrogen, we obtain benzene, which, as Kekulé demonstrated, becomes the centre round which the great group of aromatic compounds might be said to revolve. Benzene was discovered by Faraday in 1825 among the volatile liquids condensed from the oil-gas made by the Portable Gas Company. It had already played a notable part in the development of chemical theory in connection with the124 discovery of isomerism. It was now to play a far more important rôle: to become, in fact, the progenitor of a great family of substances, not only of theoretical value, but of great economic importance.
The limits of this work preclude any attempt to trace in detail the development of the conception with which Kekulé enriched science, or to dilate upon the great extension of benzenoid or cyclic chemistry which has resulted from it during the past forty years. It has been said that Kekulé’s memoir on the benzene theory is the most brilliant piece of scientific prediction to be found in the whole range of organic chemistry. Of course, on its promulgation it had to run the gauntlet of criticism; and an army of eager, active workers was soon engaged in testing its sufficiency and in developing the rich province which it first made known. As the facts multiplied, other statical formulæ were suggested by Dewar, Ladenburg, and Claus, but they have not proved more adequate to explain the facts as these have become better understood. Observations which seemed to contradict Kekulé’s theory, or which seemed to be imperfectly explained by it, have, in the light of fuller knowledge, been shown to be in harmony with it; and such additional proofs of agreement have thereby served to strengthen its position. Its capacity for development is,125 indeed, as in the case of every other hypothesis of the first rank, one of its cardinal qualities. It adequately explains the constitution of great numbers of derivatives whose analogies and relations, apart from it, would have remained obscure and in many cases unintelligible. The symmetrical distribution of the carbon and hydrogen atoms in the benzene molecule, assumed by Kekulé on indirect grounds, has been established by the independent investigations of Ladenburg and others, and its ring structure has been demonstrated by Baeyer and Perkin. Purely physical evidence, based upon its thermo-chemical and optical characters can be adduced in its support. Determinations of the molecular volume and magnetic rotation of its compounds further serve to substantiate it.

Friedrich August Kekulé was born at Darmstadt on September 7, 1829. After passing through the gymnasium of his native town, he entered, in 1847, the University of Giessen, with the intention of becoming an architect. Attracted by Liebig’s teaching, he turned to chemistry, and worked with Will on amyl sulphuric acid and its salts. In 1851 he went to Paris, heard Dumas’s lectures, and formed a friendship with Gerhardt, whose Traité de Chimie Organique largely moulded his views. He became an assistant to Von Planta, occupying himself with the chemistry of the alkaloids.126127 Subsequently he came to London, worked under Stenhouse, and made the acquaintance of Williamson, then in the full vigour of his scientific activity. Here he discovered thioacetic acid. It was at this time, also, that his ideas with regard to structural chemistry began to take shape. Returning to Germany, he attached himself to the University of Heidelberg as a privat-docent, and had for a pupil Baeyer, who took up the study of the organo-arsenic compounds. In 1858 he published his memorable paper “On the Constitution and Metamorphoses of Chemical Compounds and on the Chemical Nature of Carbon,” in which he developed his views on the linking of atoms, out of which has grown our system of constitutional formulæ. The immediate result of this celebrated memoir was a call to the chemical chair of the University of Ghent, where Kekulé had among other students Baeyer, Hübner, Körner, Ladenburg, Linnemann, and Dewar. Here he remained nine years, and here he published his classical Lehrbuch der Organischen Chemie. The years he spent in Ghent were the most productive time of his career, and it was there that he developed his benzene theory—a conception as fruitful as that of his doctrine of atom-linking. In 1867 Kekulé was called to Bonn to take charge of the newly erected laboratory which Hofmann had designed. Although he128 continued to work, mostly in collaboration with his pupils, among whom may be named Anschütz, Bernthsen, Thorpe, Carnelley, Claisen, Dittmar, Franchimont, Van ’t Hoff, Japp, Schultz, Wallach, and Zincke, his health after 1876 began to fail. He died on July 13, 1896.
Of course no statical formula can be the ultimate representation of the constitution of benzene. However convenient and suggestive such a formula may be, it can be only a transitional phase in its complete chemical and physical history. Kekulé was early conscious of this fact, and suggested a dynamical hypothesis based upon a mechanical conception of valency. This he imagined might represent the number of contacts with other atoms which a vibrating atom experienced in the unit of time. Two atoms of tetravalent carbon, each linked by one combining unit, will experience four oscillations, striking each other and three other atoms in the unit of time, while the monovalent hydrogen atom makes only one oscillation. The doubly linked carbon will collide with its neighbouring atom twice, and also with two other atoms within the same period. The assumption that the carbon atom has a more rapid motion than the hydrogen atom is, however, not warranted by the kinetic theory. Other dynamic formulæ have been proposed by Knorr and by Collie.129 Collie and Baly have further suggested that the absorption bands of benzene observed in the ultra violet of its spectrum point to synchronous oscillations of its molecule, due to dynamic changes in the making and breaking of the links between the several pairs of the carbon atoms, setting up vibrations in the benzene ring comparable with those of an elastic ring in the act of expanding and contracting.
The large group of the essential oils, containing hydrocarbons similar to oil of turpentine, and classed under the generic term of terpenes, might, from their origin and mode of occurrence, be expected to be allied in constitution to the aromatic compounds; and such is found to be the case. The terpenes are isomeric hydrocarbons of the formula C10H16. They are found sometimes singly, at other times mixed, in a great variety of plants, associated with sesquiterpenes C15H24, and oxygenated substances, such as camphor, borneol, menthol, etc., some of which have long been known and valued for their medicinal properties and technical applications. The elucidation of their constitution has taxed the skill of many workers during the past thirty years; but, thanks to the labours of Wallach, Baeyer, Perkin, Tiemann, Bredt, Komppa, and others, an insight has been gained into their nature and analogies. They are130 apparently all cyclic compounds with certain attributes which connect them with hydrocarbons of the aliphatic series. Pinene, the characteristic constituent of oil of turpentine, obtained by distilling the resinous exudations of many species of pines, exists in two modifications, distinguished by differences in optical activity, known respectively as australene, found in American, Russian, and Swedish turpentine, and terebenthene, found mainly in French turpentine. It would seem from their empirical formulæ, as well as from their association in nature, that the terpenes and camphor, which Dumas first showed to have the composition C10H16O, should be closely allied in constitution, and that it ought to be readily possible to effect their mutual transformation. The constitution of camphor was long one of the standing problems of organic chemistry, and dozens of formulæ have been suggested at various times during the last twenty years in explanation of its structure. That it contained a benzene nucleus seemed to be proved by the ease with which it yielded toluene, cymene, and other benzene homologues. The first real insight into its structure was gained when Bredt ascertained the constitution of camphoronic acid, C6H11 (CO2H)4—a product, together with camphoric acid, of the oxidation of camphor—which he found broke up into trimethylsuccinic131 acid and isobutyric acid, and the structure of which was established by Perkin and J. F. Thorpe.
The result of the Japanese monopoly has been to greatly enhance the price of natural camphor; during the last ten years it has practically trebled. This has naturally stimulated endeavours to prepare this substance by synthetical means. Artificial camphor is now made from pinene by transforming the hydrocarbon into bornyl chloride by the action of hydrochloric acid. From this camphene is prepared; by treatment with glacial acetic acid it forms isobornyl acetate. On hydrolysis this is transformed into isoborneol, which by oxidation yields camphor, differing from the naturally occurring variety only in the fact that it is optically inactive. All so-called aromatic compounds are not necessarily cyclic systems, for it has been recognised within the past few years that some of the most valuable natural perfumes, such as that of the rose, lavender, and orange blossom, lemon-grass, geranium, ylang-ylang, neroli, etc., owe their characteristic aroma to the presence of terpenes and camphors, which are not strictly benzenoid or cyclic compounds, but “ruptured rings” behaving like open-chain or aliphatic substances. To judge from past experience, it may confidently be stated that, now the constitution of these substances is132 understood, their synthetical preparation on an industrial scale is practicable. The discovery by Cahours in 1844 that oil of wintergreen is substantially methyl salicylate led to its artificial production from synthetically prepared salicylic acid. Sir William Perkin in 1868 effected the synthesis of coumarin, the aromatic principle in woodruff and hay. Fittig and Mielck in 1869 synthesised heliotropin, and in 1871 Tiemann and Haarmann obtained vanillin, the characteristic aromatic body in the vanilla pod, by synthetic means, and established its manufacture on a commercial scale. The chemical nature of the characteristic odoriferous substances in oil of cumin, anise, rue, cinnamon, heliotrope, jasmine, violet, parsley, etc., has now been established and some of them are made industrially. The artificial essence of violets known as ionone, prepared by Tiemann in 1893, and now made commercially, is similar but not identical in structure with the true perfume—irone. What is known as artificial musk is a trinitro-butyl toluol. Artificial orange-flower oil is a methyl ester of anthranilic acid.
In Vol. I. a short account has been given of the early history of the large and important group of vegetable products known as the alkaloids. Many of these have long been valued on account of their powerful physiological action. As they133 all contain nitrogen and are generally basic, they were regarded by Berzelius, and subsequently by Liebig and Hofmann, as akin to ammonia in constitution, and were classed as amines. The first experimental evidence of their nature was obtained by Gerhardt, who found that, when strychnine and certain of the alkaloids belonging to the quinine group are treated with potash, an oil was obtained which he termed quinoline, and which was recognised by Hofmann as identical with a substance obtained in 1834 from coal-tar by Runge, and at that time known as leucol. By other modes of treatment certain alkaloids—e.g., nicotine and conine—are found to yield pyridine, a basic substance found by Anderson, in 1846, in the fœtid liquor obtained by distilling bones, and since found in coal-tar. Others of them—e.g., papaverine, narcotine, etc.—yield isoquinoline, an oil also discovered in coal-tar, by Hoogewerff, and Van Dorp, in 1885. These three substances—quinoline, isoquinoline, and pyridine—constitute so many nuclei in the constitution of a large number of alkaloids. Pyridine resembles benzene in being a cyclic compound, consisting of five carbon atoms and one nitrogen atom. Quinoline stands to pyridine in much the same relation that naphthalene stands to benzene. It can be obtained synthetically, as first shown by Koenigs and Skraup, and subsequently134 by Doebner and Von Miller, from benzene derivatives.
Isoquinoline, isomeric with quinoline, differs from that substance in the position of the nitrogen atom. It, too, has been synthetically prepared from benzene derivatives in a number of ways.
Among the naturally occurring pyridine alkaloids may be named piperine, found in black pepper, and conine, the poisonous principle of hemlock (conium maculatum). The latter alkaloid was prepared synthetically by Ladenburg in 1886; as first obtained it differed from the naturally occurring product, which is dextro-rotatory, in being optically inactive. Ladenburg surmised that the synthetic preparation stood to the naturally occurring compound in the same relation that racemic acid stood to tartaric acid, and that, by treatment in the manner employed by Pasteur, the racemic modification of conine might be separated into its dextro- and lævo-constituents. This was found to be the case; but the separated dextro component was now found to be distinctly more optically active than the pure, natural variety. It was, in fact, an isomeric modification—iso-conine. By heating this to 300° it was transformed into ordinary conine, identical in all respects with the natural alkaloid. Ladenburg has also effected the synthesis of piperine135 by condensing piperidine and piperinic acid.
Nicotine, the alkaloid of tobacco, was discovered by Posselt and Reimann in 1828. Its constitution was first ascertained by Pinner, and it was synthetically obtained by Amé Pictet, in 1904, as an inactive substance, capable of being resolved by the crystallisation of its tartrates into a dextro- and lævo-modification, the latter of which was identical with that found in the tobacco leaf.
Atropine and hyoscyamine—the poisonous principles of belladonna and henbane—are isomeric alkaloids, the former of which is optically inactive, and the latter is lævo-rotatory. Atropine is, in fact, the racemic modification. The constitution of both alkaloids is known, and their synthesis is now possible.
The successive steps may be thus indicated:
1. Synthesis of glycerin (Faraday, Kolbe, Melsens, Boerhave, Friedel, and Silva).
2. Glycerin to glutaric acid (Berthelot and De Luca, Cahours and Hofmann, Erlenmeyer, Lermantoff, and Markownikoff).
3. Glutaric acid to suberone (Brown and Walker, Boussingault).
4. Suberone to tropidine (Willstätter).
5. Tropidine to tropine (Willstätter, Ladenburg).
6. Synthesis of tropic acid (Berthelot, Fittig136 and Tollens, Friedel, Ladenburg, and Rügheimer).
7. Tropine and tropic acid: atropine (Ladenburg).
The alkaloid cocaïne, contained in the leaves of erythroxylon coca and now employed as a local anæsthetic, was discovered by Niemann in 1860. It has been shown to be closely related to atropine in constitution, and has now been synthetically prepared in the dextro-modification.
The alkaloids papaverine, narcotine, narceïne, contained in opium, are derivatives of isoquinoline, as also is berberine, found in the common barberry (berberis vulgaris). Papaverine, which occurs in opium to the extent of about one per cent., was first isolated by Merck in 1848. Its constitution has been established by Goldschmidt. Narcotine is, next to morphine, the most abundant constituent of opium. The study of the products of its hydrolysis and oxidation—viz., opianic acid and cotarnine, both of which substances have long been known—has indicated its probable structure. Narceïne is closely allied to narcotine, and can, indeed, be obtained from it by combining the latter alkaloid with methyl iodide and treating the compound with caustic potash. The constitution of berberine, which is one of the few coloured vegeto-alkaloids known, has been worked out by Perkin. As yet nothing definite is known137 concerning the structure of the most important and largest constituent of opium—viz., morphine; or of its congeners codeïne and thebaine. Grimaux, however, in 1881 converted morphine into codeïne by treatment with methyl iodide and potash; hence the two alkaloids stand in a relation somewhat similar to that in which narceïne stands to narcotine. There is very little doubt that the three alkaloids are very closely related, and that a knowledge of the constitution of one of them would immediately elucidate the structure of the others. They are probably phenanthrene derivatives.
Quinine and cinchonine, the most important of the cinchona alkaloids, are quinoline compounds, and are closely related in constitution. But the many attempts to unravel their structure have yielded no definite results up to the present.
138
The first gropings in the search for light on the inner structure of molecular groupings may be said to date from Biot’s work on polarisation. In 1815 Biot, a pupil of Malus, made the remarkable discovery that a number of naturally occurring organic compounds—e.g., sugar, tartaric acid, oil of turpentine, camphor, etc., are optically active—that is, rotate the plane of polarisation in one direction or the other. The property had previously been observed in quartz, and was assumed to be connected with the crystalline character of that substance. Biot, however, pointed out that the case of oil of turpentine which is a liquid, and the cases of the other substances when in solution, showed that crystalline character had no necessary connection with the phenomenon, but that it must be dependent upon the internal or molecular arrangement of the optically active substance.
In 1844 Mitscherlich, who first demonstrated139 the relation between atomic constitution and crystalline form, drew attention to the fact that the salts of the isomeric modifications of tartaric acid, studied by Berzelius, although possessing the same chemical composition, the same crystalline form, with the same angles, the same double refraction, and therefore the same angles between their optical axes, nevertheless behave quite differently as regards their optical activity, solutions of the tartrates rotating the plane of polarisation, whereas those of the racemates are inactive. In 1848 this remarkable circumstance engaged the attention of Louis Pasteur, a young man who had just completed his course at the École Normale in Paris, and was acting as assistant to Balard, the discoverer of the element bromine. Pasteur, on examining the crystals of the two forms of tartaric acid, and of some of their salts, detected the presence, on some of them, of certain facets—so-called hemihedral faces—which had hitherto been unrecognised, but were similar to facets which Haüy had observed on quartz. Haüy had, in fact, divided quartz crystals into two classes—right-handed and left-handed, depending upon the side on which these facets occurred. The forms were, as it is termed, enantiomorphous. Biot, moreover, found that some quartz crystals, cut parallel to the axis, turned the plane of polarisation to the right, whereas others turned140 it to the left; and Herschel suggested that the phenomena were probably connected, and such was found to be the case.
Mindful of Herschel’s observation, Pasteur found that the crystals of certain of the optically active tartrates showed hemihedral faces, whereas those of the corresponding racemates showed no trace of them. On recrystallising the racemates, however, it was noticed that two sets of crystals were formed—enantiomorphic forms—the first set of crystals having hemihedral forms on the right-hand side, and the second set on the left-hand side. The forms, in fact, were so related that one appeared, as if it were the image, as seen in a mirror, of the other. When solutions of these crystals were examined, one set was found to rotate to the right, the other to an equal degree to the left. The dextro-rotatory salt yielded ordinary tartaric acid; the corresponding lævo-rotatory acid was a hitherto unknown modification: the two together, in equal proportions, constituted racemic acid.
In 1863 Wislicenus published a remarkable memoir on the synthesis of lactic acid. The acid in sour milk was discovered by Scheele in 1780. In 1807 Berzelius discovered a similar acid, called sarcolactic acid, in muscle juice; this was erroneously pronounced by Liebig to be identical with that of sour milk. Other forms of lactic acid were made known, the structural character141 of which was not to be explained by current hypotheses. Wislicenus concluded that their differences could be due only to different arrangements of their atoms in space.
In 1874 the conception of atomic grouping received a remarkable development by the publication of two memoirs—one by Van ’t Hoff, and the other by Le Bel—which served to connect molecular structure with optical activity. Confining their attention to carbon compounds, they inferred that all optically active substances contained at least one multivalent atom, united to other atoms or groups, so as to form in space an unsymmetrical arrangement. Van ’t Hoff regarded the carbon atom as occupying the centre of a tetrahedron, to the summits of which its valencies were directed. If different groupings are attached to these summits, the structure is asymmetrical, and is optically active. The two forms of lactic acid, for example, may be represented by the following space formulæ:
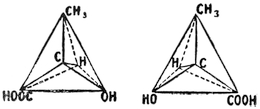
It will be seen from an inspection of the figures that the one is the image-form of the other, and,142143 no matter how they are turned, they are not superposable; they are right- and left-handed, or, as it is termed, enantiomorphs.
There is no fundamental distinction between the hypothesis of Van ’t Hoff and Le Bel as to the effect of asymmetry on optical behaviour. Le Bel regards the effect of asymmetry simply as a necessary consequence of the presence of four dissimilar groupings, and as independent of valency and the geometrical form of the molecule.

It was surmised by Pasteur that every liquid or solid in solution showing optical activity, if crystallisable, would be found to manifest hemihedral faces; but this has not been generally established. Further, it does not always happen that an optically active substance in solution is so when solid. Lastly, optical activity may be latent even in asymmetric carbon compounds if dextro- or lævo-modifications are present in equal proportions, as in racemic acid. Such compounds are, in fact, termed “racemic,” or racemoids; and they may be separated occasionally by crystallisation, as in Pasteur’s method with the tartrates; or as shown by him by the action of the racemoid upon another optically active substance; or, lastly, by taking advantage of the specific action (specific assimilation) of organisms—Pasteur’s so-called biochemical method.
144 It is a physiological fact of great interest that the behaviour of enantiomorphs towards the animal organism is frequently markedly different. Lævo-tartaric acid administered to guinea-pigs is found to be twice as poisonous as the dextro-acid; dextro-asparagine possesses a sweet taste, but lævo-asparagine is tasteless; lævo-nicotine is more poisonous than the dextro-alkaloid.
The ferments known as enzymes are also found to possess the power of selection, behaving differently towards the different optically active modifications of the same substance. It is frequently observed that an optically active substance may be rendered inactive by the conversion of half the substance into its enantiomorph. This operation was first performed by Pasteur, and may be brought about by heating the substance, either alone or with water, under pressure. Indeed, it is occasionally observed to take place at the ordinary temperature (autoracemisation).
By the action of various reagents the derivatives of an optically active substance are found not unfrequently to change the direction of their optical activity. Indeed, by such means one enantiomorph may be changed into another. Thus lævo-menthol may be converted into the dextro-modification by treatment with sulphuric acid.
The rotatory power of a substance is frequently145 modified by the character of its solvent, and varies with the temperature and concentration of the solution. Landolt and Oudemans found that the specific rotation of dilute solutions of tartrates and of salts of the active alkaloids was independent of the nature of the base and acid respectively present—a fact which finds its explanation in the theory of electrolytic dissociation. It has been known for some years past that the specific rotation of solutions of certain sugars changes with time, being sometimes less and sometimes more than the initial amount. This phenomenon is now known as multirotation, or mutarotation. It seems to be connected with an alteration in the configuration of the molecules.
There is a special case of stereo-isomerism, differing from that of optical isomerism and of structural isomerism (with which we have hitherto been alone concerned), which was predicted by Van ’t Hoff in his remarkable work La Chimie dans l’Espace, published in 1877—noteworthy as being the first serious attempt to grapple with the problem of spatial molecular grouping, foreshadowed by Wollaston, Berzelius, and, indeed, all the early philosophic thinkers who accepted the atomic theory. The special form of stereo-isomerism now referred to, which has been more particularly investigated by Wislicenus, is distinguished as geometrical146 isomerism; not, perhaps, a sufficiently descriptive term, since, comprehensively, all forms of isomerism are really cases of geometrical isomerism. Instances of it are to be met with among the isomeric acids existing as glycerides in certain fats, in cinnamic acid, in stilbene and its derivatives, etc. It was first observed in maleic and fumaric acids—isomeric acids of the empirical formula C2H2 (COOH)2, obtained by the distillation of malic acid, the characteristic acid met with in the apple and other fruits and in certain other vegetal products. These acids may be represented by the following space formulæ:
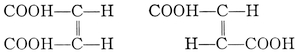
which show no asymmetry, and hence no possibility of optical activity or enantiomorphous modifications.
In the case of maleic acid it will be seen that the same groups (COOH or H) are represented on the same side of the molecule—in other words, they are placed symmetrically in a plane—whereas in fumaric acid they are placed diagonally or are axially symmetrical. Isomers of the first case are classified as malenoid or cis-forms, while those of the latter are termed fumaroid or trans-forms.
147 Substances of the character referred to are, as a rule, mutually convertible with more or less ease; they are susceptible of what is called geometrical inversion. Thus fumaric acid may be readily converted into maleic acid by heating; maleic chloride is gradually transformed into fumaric chloride at ordinary temperatures. Sunlight, or a particular solvent, or the presence of some substance which acts as a catalyst, may effect the inversion. Cis and trans isomerism is also met with among cyclic compounds; it occurs among the terpenes; and certain alkaloids, as, for example, cocaïne, exhibit it.
Although the doctrine of stereo-chemistry was first enunciated in the case of carbon, and was, indeed, for a time solely confined to compounds in which carbon was the nucleal element, there is no a priori reason why the phenomenon should be so restricted. Van ’t Hoff, in fact, in 1878, discussed the question in relation to nitrogen compounds. Stereo-isomeric nitrogen derivatives were first obtained by Victor Meyer and his pupils, and the stereo-chemistry of nitrogen has since proved to be a very fruitful field of investigation, notably in the hands of Goldschmidt, Beckmann, Hantzsch and Werner, Le Bel, Ladenburg, Bamberger, Kipping, H. O. Jones, Pope, and others. The stereo-chemistry of nitrogen differs from that of carbon, inasmuch as variation of valency plays a far more148 important part in the case of nitrogen than it has hitherto been observed to do in that of carbon; the spatial representation of the trivalent nitrogen atom differs from that of the pentavalent atom. Le Bel, in 1891, succeeded in obtaining an optically active nitrogen enantiomorph by the application of Pasteur’s biochemical method. Optically active compounds have since been prepared by Pope and Peachey and H. O. Jones. Pope and Peachey have also prepared optically active compounds of sulphur, selenium, and tin; and Kipping has obtained an asymmetric compound of silicon.
In 1863 Geuther, and, independently, Frankland and Duppa, made known the existence of aceto-acetic ester. By Geuther this compound was termed ethyl-di-acetic acid—
CH3.C(OH): CHCOOC2H5
by Frankland and Duppa it was considered to be acetone-carboxylic acid—
CH3.CO.CH2.COOC2H5.
The essential difference in these formulæ, as the two names respectively indicate, is that the first implies that the ester has an acidic or hydroxylic character, proved by its forming characteristic salts; the other that it contains the group CO,149 proved by its yielding acetone and the usual reactions of the ketones. The attempt to settle the constitution of this substance gave rise to much controversy, and, as it was found to be very reactive, led to a great amount of conflicting experimental work. The ultimate result was to show that both formulæ are correct: at the time of reaction the ester is sometimes hydroxylic, at other times ketonic, or, adopting the terminology of Brühl, it sometimes shows the enol form, at other times the keto form. Other substances were subsequently found to behave in the same way. In 1885 the question was discussed by Laar, who suggested the term tautomerism (ταὐτό, the same; μέρος, a part) to denote the fact that one and the same substance could have structural formulæ varying with conditions of reaction and depending upon the migrations of certain of its atoms within the molecule. During the last twenty years a large number of examples of the kind have been discovered. They are found to occur, not only among aliphatic substances, but in cyclic and heterocyclic compounds. We now know that such intermolecular changes may occur by the migration of any of the elements or groups present in the molecule. Thus, to confine ourselves to simple and well-known examples, the transformation of sodium phenyl carbonate into sodium salicylate, discovered by Kolbe, is due150 to the wandering of an atom of hydrogen from the benzene residue to oxygen, thus:
The conversion of the nitriles into the cyanides by heating is due to the transference of the alkyl radical from the nitrogen atom to the carbon—
R.NC➞NC.R.
Alkyl groups may also be transferred from oxygen to nitrogen; a radical may detach itself from a carbon atom and wander to a nitrogen atom; radicals in cyclic compounds may be transferred from the side chains to the nucleus, etc.
The phenomenon, in fact, is now so general that grave doubts have been thrown upon the uniform value of deducing the structural formula of a substance from the study of its decomposition products, or from the nature of its derivatives, owing to the readiness with which tautomerism may occur. The change may be brought about by variation of temperature, by the reagent itself, by the action of a solvent or the presence of a catalyst—that is, of a substance which apparently plays no part in the metamorphosis. Hence the value of specific reagents as clues to constitution is considerably weakened, since151 the results may be equivocal. Fortunately, the great extension, within recent years, of the application of physical methods has considerably strengthened our means of gaining an insight into molecular structure; and the investigations of Brühl on refraction and dispersion, of Perkin on magnetic rotation, of Hantzsch on electrical conductivity, of Lowry on solubility, of Lowry and E. F. Armstrong on optical activity, of Knorr and Findlay on melting-points, and, lastly, of Hartley, Dobbie, Lauder, Baly, and Desch on absorption spectra, have collectively afforded valuable information on the mechanism of isomeric change based upon dynamical considerations.
Space will not permit of a more extended treatment of the subject of stereo-chemistry; and certain matters relating to it, as, for example, the phenomena classed under the term steric hindrance, must be left unnoticed. This term has reference to the hindrance which certain groups, or the particular distribution in space of certain atoms, exert on the progress or extent of a reaction, as, for example, of hydrolysis or esterification, etc. The influence of special groupings in retarding chemical change is apparently well established, but no comprehensive theory of the subject is yet possible. Until such a theory is forthcoming a dynamical theory of stereo-chemistry is incomplete.
152
In its widest sense, the term “synthesis,” as used in organic chemistry, means the building-up of carbon compounds, either from their constituent elements or from groups of differently constituted molecules. At one period this term was confined to cases in which the organic compound was prepared from inorganic materials, or from combinations which themselves could be formed from their elements; but latterly it has lost, in large measure, this restricted signification. At the same time, the attempt has been made to indicate by special terms certain classes of synthetical reactions. Thus the special case of the formation of an organic compound by the union of two or, it may be, more molecular groupings is now frequently spoken of as condensation.
Organic chemistry has been largely developed by the discovery from time to time of special reagents and special types of reactions which153 have shown themselves to be capable of extensive application. Such, for example, was Frankland’s discovery, in 1852, of zinc-ethyl—the first of the organo-metallic compounds, and the type of a series of substances of great theoretical importance, and of great practical value by reason of their reactive powers. They led to the synthesis of the paraffins, the secondary and tertiary alcohols, and ketones. A few years later Wurtz introduced the use of metallic sodium as a condensing agent, and showed thereby how the hydrocarbon butane could be produced from ethyl iodide:
Use was made of the same agent by Fittig, in 1863, in effecting the synthesis of the homologues of benzene by the action of an alkyl iodide upon bromobenzene:
In like manner Kekulé, in 1866, obtained benzoic acid by the action of carbon dioxide upon bromobenzene:
The readiness with which magnesium can now be obtained, mainly as the result of Sonstadt’s154 efforts to develop its metallurgy, has led to its application, at the suggestion of Barbier, in 1899, in place of zinc. The particular form of magnesium compound now employed as a reagent was prepared by Grignard in 1900, and is known by his name. It is obtained by bringing an ethereal solution of an alkyl iodide or bromide into contact with magnesium, when the metal is dissolved, forming, in the case of methyl iodide,
Grignard’s reagent has shown itself to be extraordinarily reactive, and a great number of condensations—of hydrocarbons, alcohols, aldehydes, acids, ketones, amides, and additive compounds—have been effected by means of it.
Other condensing reagents of value are aceto-acetic ester, sodium amalgam, sodamide, sodium ethoxide, dimethyl sulphate, zinc chloride, aluminium chloride, fused caustic potash, hydrogen chloride, phenyl-hydrazine, hydrogen peroxide in presence of a ferrous salt (Fenton’s reagent), ammonia, and various amines. The application of these reagents has led to the discovery of a variety of new compounds, the mode of origin of which has served to elucidate their constitution.
The great majority of organic syntheses, especially when they start by the use of inorganic155 materials, consist in passing from simple to complex molecular groupings by condensation processes. An interesting example of the reverse process is seen in the production of carbon suboxide, or carbon carbonyl, C3O2, obtained from various malonyl compounds, but most conveniently prepared by the action of phosphoric oxide on malonic acid under diminished pressure, or by treating an ethereal solution of dibromomalonyl chloride with zinc:
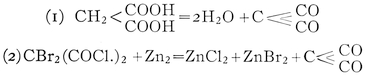
Carbon suboxide is a colourless, extremely mobile, refractive, poisonous liquid, of sp. gr. 1.11, with a strong and peculiar smell. It boils at 7°, and freezes at -107°. It is stable only at low temperatures; at ordinary temperatures it polymerises to a red solid, which dissolves in water, forming a solution of the colour of eosin. The change is almost instantaneous at 100°. Carbon suboxide is inflammable, burning with a blue but smoky flame: C3O2 + 2O2 = 3CO2. Its low boiling-point and the high value of its molecular refraction and dispersion, its general resemblance to the metallic carbonyls and ketones, etc., indicate that this remarkable156 oxide of carbon is, in all probability, the anhydride of malonic acid. Indeed, by the action of water upon it, it is reconverted into malonic acid.
In point of principle, and viewed as chemical operations, the synthesis of vital products is in nowise different from the synthesis of any other group of organic compounds; and the special interest, and even astonishment, at one time created by the artificial preparation of such products has largely died away. The synthetical production of some of the substances formerly known only to be formed by vital action, either in the animal or the plant, has already been incidentally referred to. But it may be convenient to treat the subject of the artificial production of this group of bodies rather more comprehensively and as a sub-section of this chapter on organic synthesis, since their formation by such means constitutes a phase in the development of chemistry, and has undoubtedly exercised a profound influence on scientific thought and on philosophical and even theological doctrine. During the past fifty or sixty years the chemist has been enabled to form the active principles or characteristic products of many plants and animals. He has built up substances which were formerly regarded as capable of being made only by the very process of living. He has prepared compounds which157 were at one time considered as only producible by changes in organised matter after death.
Since the date of Wöhler’s epoch-making discovery, already referred to4 urea has been synthetically prepared by many reactions, notably by Regnault and Natanson by the action of ammonia on carbonyl chloride, and by Basarow and Dexter from ammonium carbamate. Both these substances can be formed directly or indirectly from their elements. It may also be obtained by the hydrolysis of lead cyanate:
4 Vol. I., p. 163.
The successive steps in its production from inorganic materials by this method are:
Associated with urea as products of metabolism are uric acid, xanthine, and sarcine. Urea was first artificially transformed into uric acid by Horbaczewski, and its synthesis was effected by Behrend and Roosen. Closely related in chemical composition to these substances are theobromine and caffeine, the characteristic principles respectively of cocoa (the fruit of theobroma cacao); and of coffee, tea, maté (the leaves of ilex paraguayensis); “guarana,” obtained from the seeds of the South American158 plant paullinia sorbilis, and the kola-nut of Central Africa. Strecker, in 1860, showed how theobromine may be converted into caffeine; and Emil Fischer, by similar means, transformed xanthine into theobromine. Since that time xanthine itself has been prepared artificially. Caffeine can now be built up from its elements by a series of transformations effected by a succession of chemists, as follows:
1. Carbon and oxygen give carbonic oxide.—Priestley, Cruickshank.
2. Carbonic oxide and chlorine give carbonyl chloride.—J. Davy.
3. Carbonyl chloride and ammonia give urea.—Natanson.
4. Urea gives uric acid.—Horbaczewski; Behrend and Roosen.
5. Uric acid gives xanthine.—E. Fischer.
6. Xanthine gives theobromine.—Strecker.
7. Theobromine gives caffeine.—E. Fischer.
Synthetic theobromine is now made on the large scale, and introduced as a soda compound, in combination with sodium acetate, into medicine as a diuretic under the name of agurin. Synthetic caffeine is also prepared on a manufacturing scale from uric acid through the medium of the methylxanthines. The close relationship of xanthine to uric acid is of great159 physiological significance, since there is little doubt that the xanthine bases are the most important sources of uric acid within the organism.
In this connection reference may be made to the large number of synthetic organic products which have been introduced into medicine during the past few years. The investigation of the constitution of the alkaloids has served to show in many cases to what particular molecular grouping the physiological action of the drug is mainly due, and this has led to the production of substances containing these groups, but not necessarily existing as natural products. Among these may be mentioned antipyrin, a derivative of the pyrazol group, discovered by Knorr in 1883, and of which upwards of 17,000 kilos, of the approximate value of £35,000, were produced in 1899. This substance is a phenyl-dimethyl-pyrazolone.
Acetanilide C6H5NH.COCH3, an aniline derivative, was discovered by Gerhardt in 1853. Phenacetin is a derivative of para-aminophenol:
An extraordinary number of synthetical soporifics have been introduced at various times during recent years—e.g., chloral hydrate, veronal, sulphonal, trional and tetronal, etc. The three160 last-named substances are closely related, as the following formulæ indicate:
Sulphonal is prepared by the oxidation of a substance obtained by the combination of acetone and ethylmercaptan. Veronal is an ethyl compound of barbituric acid, obtained by the condensation of urea and diethyl malonyl chloride:
Attempts have been made to connect the physiological working of local anæsthetics with particular constitutional groupings, as, for example, in cocaïne; and these have led to the introduction of such substances as the orthoforms, nirvanine, stovaïne, alyhine, novocaïne, and adrenaline into medicine. Adrenaline, used in conjunction with cocaïne, has proved itself a most valuable agent in producing what is called lumbal anæsthesia, whereby large sections of the161 lower half of the body may be rendered completely insensitive to pain.
The study of the putrefactive changes of albuminous substances of animal origin, induced by the activity of micro-organisms, has revealed the existence of a number of basic nitrogenous compounds, some of which are highly poisonous. These were classed by Selmi under the generic name of ptomaines (πτῶμα, a corpse). Brieger found that the typhoid bacillus yielded a poisonous substance—typhotoxine, and that the bacillus of tetanus forms a highly toxic basic body, tetanine. All the ptomaines, however, are not poisonous. Some of them, like choline (χυλὴ, bile)—originally discovered by Strecker in bile, in the brain, in yolk of egg, and now found to be among the products of the putrefaction of meat and fish—have been known for some time past. Choline was first synthetically prepared by Wurtz. Neurine (νεὺρον, nerve), a derivative of brain substance, is related to choline, and is readily transformed into it, but differs from it in being very poisonous. It has been synthesised by Hofmann and by Baeyer. Another of the so-called corpse-alkaloids—cadaverine—has been synthetically formed by Ladenburg. Schmiedeberg and Kopp isolated the poisonous principle of the fungus agaricus muscarius, which they named muscarine. It occurs with choline, from which it can be162 readily obtained, among the products of the putrefaction of flesh, as well as in many fungi.
The synthesis of the alkaloids conine, atropine, cocaïne, piperine, and nicotine has been already referred to5 as also that of vanillin, the aromatic principle of the dried fermented pods of certain orchids; coumarin, the odoriferous principle of woodruff and of the tonka bean; of salicylic acid, oil of wintergreen, oil of mustard, bitter-almond oil, and camphor. Acetic, succinic, tartaric, and citric acids have also been artificially obtained, and may, indeed, be built up from their elements.
5 P. 133.
No synthesis of recent years created more widespread interest than that of alizarin, first effected by Graebe and Liebermann in 1868. Its successful commercial manufacture by Sir William Perkin in this country and by Caro in Germany created nothing less than a revolution in one of our leading industries, and completely destroyed a staple trade of France, Holland, Italy, and Turkey. To procure alizarin, anthraquinone is treated with sulphuric acid, and the product is fused with alkali and potassium chlorate.
The remarkable industrial results attending the synthetical formation of this madder-product naturally led to attempts to procure other important vegetable dye-stuffs artificially, notably indigo. The synthetical production of163 indigo has been accomplished by the joint labours of many chemists, notably Baeyer, Heumann, and Heymann, and the substance is now prepared on an industrial scale. The starting-point is naphthalene, obtained from coal-tar. This is converted into phthalic acid, which is then transformed into phthalimide. The last-named substance is converted into anthranilic acid, which, on treatment with monochloracetic acid, is changed into phenylglycin-ortho-carbonic acid. On melting this with caustic potash it yields indoxyl acid, which is transformed into indoxyl, and thence into indigo.
Another method is to treat the sodium salt of phenylglycin with sodamide, whereby indoxyl is at once obtained, and this by condensation yields indigo blue:
Phenylglycin is obtained by the action of monochloracetic acid on aniline, which in its turn is164 obtained through nitrobenzene from benzene. Since benzene can be synthetically prepared by the condensation of acetylene, which can be obtained by the direct union of carbon and hydrogen at a high temperature, it is theoretically possible to build up indigo blue from inorganic materials.
Synthetical indigo blue was placed on the market in 1897 with an almost immediate effect on the production and price of the natural variety, and to-day the output of Bengal indigo has fallen by more than fifty per cent. In 1902 the amount of the natural product was probably not greater than three million kilos, whereas in the same year the production of synthetic indigo was upwards of five million kilos. Before the introduction of the artificial variety the price of pure indigo blue ranged from sixteen to twenty shillings per kilo; by the end of 1905 it had fallen to seven or eight shillings. Mention should be made also of thio-indigo red and the thionaphthene derivatives, some of which promise to be important colouring matters. In recent years the so-called sulphur colouring matters have acquired considerable importance. Space will not permit of any fuller treatment of the development of the manufacture of the artificial organic colouring matters. This industry had its beginnings in England, but it is now mainly carried on in Germany. Its importance may be165 gleaned from the fact that the value of the production at the present time amounts to not less than £12,500,000 per annum, two thirds of the output being exported. It demands the services of battalions of skilled chemists, and gives employment to many thousands of artisans.
Some of the most notable achievements of modern synthetical chemistry are to be found in the work of Emil Fischer on the sugars and the proteins. Although the sugars have from the earliest times been reckoned among the most characteristic products of plant life, and have long been used as food and as sources of alcohol, comparatively little was known until lately of their real nature and mutual relations, in spite of numerous attempts to elucidate their constitution. Much of the mystery surrounding their chemical history has now been dispelled. Not only has the molecular structure of the more important naturally occurring sugars been unravelled, but a large number of hitherto unknown members of the various groups of the great family to which they belong have been prepared. The first insight into the constitution of these bodies may be said to date from the researches of Kiliani, made about a quarter of a century ago. In 1887 Fischer effected the synthesis of a form of fructose (fruit sugar), and immediately afterwards of ordinary dextro-glucose (grape sugar) and its enantiomorph166167 lævo-glucose, and the two optically active forms of natural fruit sugar. Since that time such sugars as arabinose, xylose, fucose, mannose, sorbose, cane-sugar, maltose, lactose, etc., and the sugars existing as glucosides, have been examined, their stereo-chemical relations defined, and synthetic methods of production devised. Incidentally, their behaviour towards enzymes has been studied, and the remarkable selective action of these ferments on the various groups, due apparently to differences of configuration, has been established, with the result that much light has been thrown on the mechanism of enzyme action and on the general theory of fermentation.

The study of the proteins by Fischer constitutes a new chapter in bio-chemistry. Although long recognised as among the most important of vital products, from the circumstance that they enter into the composition of animal tissues and secretions and are essential constituents of protoplasm, the proteins are among the worst defined substances known to the chemist. They are difficult to separate, as they closely resemble one another, and afford no certain indications of individuality. Very few of them have been obtained in a form in which their identity could be established. Oxyhæmoglobin was isolated some years ago, but the proteins of serum albumin and of egg albumin168 have only recently been obtained in definite crystalline shape. All the proteins—even the simplest of them—are of great complexity, and possess apparently very high molecular weights. Hæmoglobin, for example, appears to have approximately the formula C158H123N195O218FeS3, with a minimum molecular weight of 16,600. Indeed, there is experimental evidence to show that it is even considerably higher than this.
The main clues to the nature of these substances have been gained by the systematic study of their hydrolysis, induced by reagents, or by the action of enzymes, whereby they are found to break down into proteoses, peptones, and a great variety of amino acids, some of which have been synthesised. Among the proteins of simplest constitution are the protamines, found in the spermatozoa of fish. They are basic substances, especially rich in nitrogen, forming salts with platinum chloride and certain metallic oxides. The best investigated member of the group is salmine, obtained from the testicle of the salmon. The products of its hydrolysis have been fairly well ascertained, and their quantitative relation is such that the substance must have at least a molecular weight of 2045, corresponding to the formula C81H155N45O18. Many of the albumins and globulins—coagulable proteins contained in the animal tissues—have been isolated169 in a more or less definite form, and some of them have been found to yield substances akin to carbohydrates. Thyreoglobulin, the globulin of the thyroid gland, has been found to contain iodine, apparently as a normal constituent of a body which can be isolated as a definite proximate principle. The presence of this element is possibly connected with the curative value of the globulin in crétinism. A considerable amount of work on the vegetable albumins has also been done of late years; and some of them, as edestin from hemp seed and zein from maize, have been obtained in definite form.
The limits of this work preclude a more detailed account of one of the most interesting but at the same time one of the most obscure departments of chemistry. The field has hitherto been tilled in a somewhat intermittent and partial manner. Now that it has been entered by chemists of experience and resourcefulness, armed with modern methods of cultivation, it will doubtless soon yield a rich harvest of facts, valuable alike to the physiologist and the physician.
There can be no reasonable doubt that the chemical processes of organic life are essentially similar to those of the laboratory. The doctrine that a special “vital force” is concerned in the production of vital products receives no support170 from the teaching of modern science, and is, indeed, contradicted by it. At the same time, it must be admitted that we know very little as yet of the real agencies at work in the elaboration and mutations of chemical products in the living organism. Because we have effected the putting together of such a product by purely laboratory processes—it may be, indeed, by a variety of different and dissimilar processes—it by no means follows that any one of them is identical with that actually occurring in nature. The building up of materials in the plant by the agency of light, for example, has not yet been imitated in the laboratory. Many plant products are produced by the action of unorganised ferments—so-called enzymes—none of which the chemist has succeeded in creating.
Processes akin to condensation undoubtedly occur in the living organism; but the means by which they are effected are, in all probability, very different from anything known to the chemist at present. Many laboratory condensations are only accomplished at relatively high temperatures or under considerable pressure—or, in other words, under totally different conditions from those which obtain in the organism.
171
Chemistry and physics are each complementary to the other: that region of inquiry in which they mutually overlap is known as physical chemistry. Its beginnings are practically contemporaneous with those of chemistry itself. Its main development has occurred, however, during the last twenty-five years. Certain of its leading features have been referred to already in connection with the establishment of the fundamental principles of chemistry, the explanation of the so-called gaseous laws, the constitution of gases, the relations of their volumes to heat and pressure, and the conditions affecting their transition to the liquid state.
As regards the molecular volumes of gases it has been shown that simple relations are obtained when quantities represented by their respective molecular weights are compared under identical conditions of temperature and pressure—that is,172 under circumstances in which equal numbers of molecules form the basis of comparison. The investigation of the molecular volumes of liquids is complicated by the uncertainty as to what constitutes in their case a valid condition of comparison. Kopp’s assumption that a comparable condition was the temperature at which the vapour pressures of the liquids are equal to the mean atmospheric pressure was justified by the fact that the boiling-points of liquids are approximately two thirds of their respective critical temperatures. His conclusions have been confirmed and extended by Lossen, Thorpe, and Schiff. It has been shown that the molecular volume of a liquid—that is, the product of its relative density at the boiling-point into its molecular weight—is in the main an additive function modified by constitutive influences. Definite values have thus been obtained for a number of the elements from a comparison of homologous or similarly constituted compounds; and in certain cases these are found to be practically identical with the values of the elements in the uncombined state.
Considerable light has been gained during the last two decades concerning the nature of solution. In its most comprehensive sense solution means the homogeneous mixture of two or more substances: thus the gases which exert no chemical action on each other are mutually soluble;173 gases, liquids, and solids may be soluble in liquids; and, lastly, solids maybe soluble in solids, forming what are known as solid solutions. The mutual solubility of gases was studied by Dalton who enunciated the law of partial pressures, which states that the total pressure of a mixture of gases is the sum of the pressures exerted by the individual components. This, like all the so-called gaseous laws, is necessarily not strictly accurate under ordinary conditions, but approximates to truth in proportion as the gases are rarefied. Van ’t Hoff pointed out that the true partial pressures of the components of a gaseous mixture might be experimentally ascertained by the use of a membrane capable of effecting their separation, and on this principle Ramsay measured the partial pressures of a mixture of hydrogen and nitrogen contained in a palladium vessel connected with a manometer. The palladium, at a sufficiently high temperature, is permeable to hydrogen to the exclusion of the nitrogen. The conditions affecting the solubility of gases in liquids were experimentally studied by Dalton and Henry, and what is known as Henry’s law implies that the volume of a gas dissolved by a definite volume of a liquid is independent of the pressure; or, in other words, the density (concentration) of the gas in solution is proportional to that in the space above the liquid. Gases are dissolved by174 liquids in very different amounts, but nothing definite is known as yet concerning the relation between the nature of the gas and its solubility, although certain broad generalisations are possible. Thus neutral gases—e.g., hydrogen and nitrogen—are sparingly soluble, whereas gases which show acidic or basic properties, such as the hydrogen halides, etc., ammonia, etc., are freely soluble. Easily liquefiable gases are also comparatively soluble as noted by Graham.
Comparatively little is known definitely concerning the conditions of solubility of liquids in liquids. Some liquids are wholly, others partially miscible; and temperature and pressure appear to affect the proportions in which the components form a homogeneous mixture. As regards the solubility of solids in liquids, our knowledge is more extensive, and a considerable body of literature exists on the subject, chiefly concerning solubility of solids in water. The solubility of a solid depends on the temperature of the solvent, and, as a rule, increases with the temperature until a certain amount of the solid has been dissolved, when the solution is said to be saturated. If the clear saturated solution be slowly cooled, say, to a particular temperature, it is frequently observed that more of the solid remains in solution than is normal to that temperature; such a solution is said to be supersaturated.175 On adding some of the solid to the supersaturated solution the excess of the solute is precipitated. In certain cases of solubility of substances in water, increase of temperature appears to diminish the amount dissolved. In nearly all such cases the difference in solubility is due to differences in the hydration of the solute. The phenomena of solid solutions have been less perfectly investigated, but the facts appear to show that such solutions in general tend to obey the laws regulating the solution of liquids in liquids. Alloys may be looked upon as solid solutions; and Roberts-Austen has shown that metals are capable of intradiffusion, like liquids and gases respectively.
The general question of solution was greatly developed in 1885 by Van ’t Hoff, by specially considering the case of dilute solutions. The gaseous laws are capable of their simplest expression when the gases are rarefied to such an extent that their molecules exert no sensible mutual influence. The case of dilute solutions is analogous. If the solute is present only in very small amount, the mutual influence of its molecules is practically negligible. Under such conditions it obeys the laws hitherto supposed to be applicable only to matter in the gaseous state.
It may be desirable to explain how this fundamental fact was recognised. It has long been176 known to the physiologist that certain membranes are semi-permeable—that is, they allow of the passage of certain liquids, and of substances in solution, to the exclusion of others. This phenomenon is termed osmosis, and is of great biological significance. It was first studied by plant-physiologists, notably by Traube and Pfeffer. Many such semi-permeable membranes can be formed artificially, but the most generally convenient is found to be one consisting of copper ferrocyanide deposited on the walls of a porous vessel.
If a vessel so prepared be filled with a solution of sugar, and be then placed in water, the water is found to pass through the membrane, but the membrane is impermeable to the sugar. In consequence pressure, termed osmotic pressure, is found to occur within the pot, and may be measured by suitable means. These osmotic pressures may at times be very large: thus a 1 per cent. solution of sugar may exert a pressure of half an atmosphere, and in the case of a solution of potassium nitrate of the same concentration it may amount to a couple of atmospheres.
Pfeffer determined the relation of the osmotic pressures to the concentration of solutions of these substances, measuring the pressures in centimetres of mercury by a manometer attached to the closed porous vessel. His results in the case of sugar were as follows:
177
| Percentage strength (C). | Pressure in cm. of mercury (P). | P/C. |
|---|---|---|
| 1 | 53.5 | 53.5 |
| 2 | 101.6 | 50.8 |
| 4 | 208.2 | 52.1 |
| 6 | 307.5 | 51.3 |
It will be seen from these numbers that the ratio P/C is practically constant—that is, the osmotic pressure varies directly as the concentration. It was further found that the osmotic pressure exerted by a solution of uniform strength increases with the temperature.
The importance, of these observations in relation to the general theory of solution was first recognised by Van ’t Hoff. Osmotic pressure was regarded by him as analogous to gaseous pressure. Since P/C is constant for any one substance, and since for a definite weight of the solute the concentration is inversely as the volume of the solution, we obtain an equation analogous to the statement of Boyle’s law, PV = constant. Van ’t Hoff also found that the osmotic pressure is proportional to the absolute temperature, like the gaseous pressure. From these results, in conjunction with Avogadro’s hypothesis, it follows that the osmotic pressure exerted by any substance in solution is the same as it would exert if present as gas in the same volume as that occupied by the solution, provided that the178 solution is so dilute that the volume occupied by the solute is negligible in comparison with that occupied by the solvent. Another important consequence is that solutes, when present in the ratio of their molecular weights in equal volumes of the same solvent, exert the same osmotic pressure. Such solutions are said to be isomotic or isotonic. It can be proved by thermodynamical reasoning that depression of the vapour pressure and freezing-point of a solution is proportional to its osmotic pressure. The significance of this relation in connection with the determination of the molecular weight of a soluble substance has already been referred to.6
Determinations of molecular freezing-point depressions by Raoult and others showed that certain substances exerted only about half the osmotic pressure calculated from their known formulæ, whereas others have abnormally high osmotic pressures. The explanation of the discrepancies in the latter case was given in 1887 by Arrhenius, who pointed out that only those solutions which have abnormally high osmotic pressures are electrically conductive. This pregnant observation proved to be very fruitful in suggestiveness; and the connection between conductivity and Van ’t Hoff’s theory of solution was developed by Arrhenius into the doctrine of electrolytic dissociation or ionisation—one of179 the most important consequences of Faraday’s electrolytic laws, the work of Hittorf, and the kinetic conceptions of Williamson and Clausius to which the last quarter of a century has given rise. Arrhenius showed that not only were free ions present in an electrically conductive solution before electrolysis, as maintained by Clausius, but that the proportion of molecules dissociated into ions could be calculated from measurements of electrical conductivity, as well as from measurements of osmotic pressure. Both methods give concordant results—a strong confirmation of the validity of the theory. In a solution of common salt, containing a gramme equivalent of that substance in a litre, Arrhenius calculated that only about three tenths of the salt exists as NaCl, the remaining seven tenths being resolved into independent ions of chlorine (chloridion) and sodium (sodion): NaCl⇄[Na·] + Cl´, each moving freely in all directions, like gaseous molecules. On passing the current, electrodes placed in the solution exert a directive action on the free ions, these alone being concerned in determining the conductivity, the un-ionised molecules or the solvent itself exercising no influence. Methods of determining the migration velocity of the ions have been worked out by Hittorf, Kohlrausch, Lodge, and others.

The theory of ionisation affords a satisfactory180181 explanation of many chemical phenomena. It accounts for the characteristic properties of acids, and explains why different acids have varying “strengths” and why a “weak” acid has the same “strength” as the “strong” acid at high equivalent dilutions: in each case the acid is nearly completely ionised—in other words, the “strength” of an acid depends on the concentration of its hydrogen ions. So, too, the “strength” of a base is related to the number of its hydroxyl ions. Aqueous ammonia is relatively a “weak” base—its solution contains few hydroxyl ions. On the other hand, caustic potash is a “strong” base—its solution, on moderate dilution, is almost completely ionised: KOH = K· + OH´, the positive ion being represented by one or more dots, and the negative ion by one or more dashes. The theory accounts, too, for many phenomena in analytical chemistry—such as why magnesia is precipitated by ammonia only in the absence of ammonium chloride, and why sulphuretted hydrogen throws down zinc sulphide in the absence of hydrochloric acid. It also serves to explain many thermo-chemical facts observed by Hess, Thomsen, and others, such as the fact that the heat of neutralisation of the “strong” acids and bases is independent of their nature, and has the uniform value of 13,700 calories, in agreement with the value, as calculated by 182Van ’t Hoff, for the reaction H· + OH´ = H2O, deduced from Kohlrausch’s measurements of the conductivity of water at varying temperatures.
Certain phenomena relative to the effect of concentration (mass action) in determining chemical change—many of which have been studied by Ostwald and his pupils, as, for example, why two dilute solutions can be mixed together without thermal disturbance; numerous hydrolytic actions; the alkalinity and acidity of salts on solution; the behaviour of the “indicators” in analysis; such phenomena as the precipitability of common salt in aqueous solution by hydrogen chloride; the influence of an excess of a precipitant; the varying behaviour of reagents; the varying colour of salt solutions; the reason why water is formed in so many reactions; why a potential difference occurs at the surface of two electrolytic solutions, etc.—phenomena for the most part otherwise unintelligible, are all capable of explanation by means of it.
Although, in the above statement, we have been mainly concerned with aqueous solutions, it should be said that the theory of ionisation is applicable to other solvents, organic and inorganic. Moreover, it should be added, the theory has not been universally accepted as accounting for all the phenomena of solution. Many substances form definite hydrates which183 can be isolated, and it is a moot point whether such hydrates are capable of existing in aqueous solution, as contended by Mendeléeff, Pickering, Kahlenberg, Armstrong, and others. Such hydrates are, however, unstable compounds, affected by temperature changes, and dissociable on dilution in accordance with the law of concentration (mass action). Further, there is evidence, largely based on the work of Kohlrausch, H. C. Jones, and Lowry, to show that the ions in aqueous solutions of electrolytes are themselves hydrated.
Limitations of space preclude further attempts to deal with the development of physical chemistry during the last half-century, and many important matters must remain practically unnoticed.
The subject of thermo-chemistry is mainly the creation of the last half-century, elaborated by the labours of Hess, Andrews, Thomsen, Favre and Silbermann, and Berthelot. The work of Wenzel and Berthollet on the influence of molecular concentration on chemical change has been greatly extended by Berthelot, Guldberg and Waage, Julius Thomsen, Van ’t Hoff, Harcourt and Esson, and Le Chatelier; and the theory of mass action and the nature of reversible processes are now capable of definite expression, and can be proved independently by thermo-dynamical and kinetic reasoning.184 The phenomena of catalysis and the action of enzymes and of fermentation in general have received attention from many investigators. The phenomena of gaseous transpiration have been studied by Graham, Maxwell, and O. E. Meyer. Thermal dissociation has been experimentally observed by Deville, Troost, and others, and mathematically investigated by Willard Gibbs and Van der Waals; and its analogy to electrolytic dissociation has been established. The nature of gaseous explosions has been investigated by Berthelot, Le Chatelier, Abel, and Dixon. Important work has been done by Gladstone, Lorentz, Landolt, Nasini, Brühl, and others, on the connection between the nature and constitution of substances and their optical characters. Similar work has been done by Sir William Perkin as regards their magnetic rotation, and by Thorpe and Rodger with reference to their viscosity. The theory of phases, originating with Gibbs and developed by Van der Waals and Roozeboom, has been greatly extended. Sir J. J. Thomson and Sir J. Larmor have elaborated an electrical theory of the atom. Barlow and Pope have traced the relation between valency and volume, and the accurate measurements of Groth and of Tutton have extended our knowledge of the crystallographic relations of correlated substances.
185 Lastly, the whole subject of photo-chemistry, although originating with the observations of Ingenhousz, Scheele, and Senebier, may be said to have been studied only within our own time, notably by Bunsen and Roscoe, Pringsheim, Pfeffer, Vogel, and Abney.
Relating to the Period Covered by Vol II.
Alembic Club, Publications of the. W. Clay, Edinburgh.
Arrhenius, Svante. Theories of Chemistry. Translated by T. Slater Price. Longmans, 1907.
Beilstein’s Handbuch der Organischen Chemie. Nine vols. Leopold Voss, Hamburg, 1901–1906.
Bischoff’s Materialen der Stereochemie. Vieweg and Son, Brunswick, 1904.
Bischoff and Walden’s Handbuch der Stereochemie. H. Beckhold, Frankfort-on-the-Main, 1894.
Cain, J. C., and Thorpe, J. F. Synthetic Dyestuffs and Intermediate Products. Griffin and Co., 1905.
Chemical Society’s Annual Reports. Gurney and Jackson.
Chemical Society. Memorial Lectures, 1893–1900. Gurney and Jackson, 1901.
Cohen, Julius B. Organic Chemistry. Edward Arnold, 1907.
188 Curie, Marie. Radio-Active Substances. “Chemical News,” London, 1903.
Findlay, A. The Phase Rule. Longmans, 1904.
Fischer, Emil. Die Aminosäuren, Polypeptide und Proteine. Julius Springer, Berlin, 1906.
Fischer, Emil. Untersuchungen in der Puringruppe. Julius Springer, Berlin, 1907.
Fischer, Emil. Untersuchungen über Kohlenhydrate und Fermente. Julius Springer, Berlin, 1909.
Freund, Ida. Study of Chemical Composition. Cambridge University Press, 1904.
Garrett, A. E. The Periodic Law. Kegan Paul, 1909.
Ladenburg, Albert. Development of Chemistry since the Time of Lavoisier. Translated by Leonard Dobbin. Alembic Club, Edinburgh, 1900.
Landolt, H. Optical Activity and Chemical Composition. Translated by J. McCrae. Whittaker, 1900.
Laurent, A. Chemical Method. Translated by W. Odling. Cavendish Society’s Publications, London, 1855.
Mann, Gustav. Chemistry of the Proteids. Macmillan, 1906.
Maxwell, Clerk. Theory of Heat. With corrections and additions by Lord Rayleigh. Longmans.
189 Meldola, Raphael. Chemical Synthesis of Vital Products. Edward Arnold, 1904.
Mendeléeff, D. Principles of Chemistry. Translated by Kamensky and Greenaway. Longmans.
Meyer, Lothar. Outlines of Theoretical Chemistry. Translated by Bedson and Williams. Longmans, 1892.
Meyer, Lothar. Modern Theories of Chemistry. Translated by Bedson and Williams. Longmans, 1888.
Meyer and Jacobson’s Lehrbuch der Organischen Chemie. Veit and Co., Leipzig.
Meyer, O. E. The Kinetic Theory of Gases. Translated by R. E. Baynes. Longmans, 1899.
Muir, M. M. Pattison. History of Chemical Theories and Laws. John Wiley and Sons, New York, 1907.
Nernst, Walter. Theoretical Chemistry. Translated by C. S. Palmer. Macmillan, 1895.
Ostwald’s Klassiker der Exakten Wissenschaften.
Pictet, Amé. Vegetable Alkaloids. Translated by H. C. Biddle. John Wiley and Sons, New York, 1904.
Richter’s Lexikon der Kohlenstoff Verbindungen. Leopold Voss, Hamburg and Leipzig.
Roscoe and Schorlemmer, Treatise on Chemistry. Macmillan.
190 Rutherford, E. Radio-Activity. Cambridge University Press, 1904.
Schorlemmer, Carl. Rise and Development of Organic Chemistry. Edited by Arthur Smithells. Macmillan.
Schryver, S. B. Chemistry of the Albumens. Murray, 1906.
Soddy, F. Radio-Activity. The Electrician Printing and Publishing Co., London, 1904.
Stas. Recherches sur les Rapports réciproques des Poids Atomique, 1860–1865.
Stewart, A. W. Recent Advances in Organic Chemistry. Longmans, 1908.
Stewart, A. W. Stereo Chemistry. Longmans, 1907.
Thorpe, T. E. Dictionary of Applied Chemistry. Three vols. Longmans.
Thorpe, T. E. Essays in Historical Chemistry. Macmillan, 1902.
Travers, Morris W. Study of Gases. Macmillan, 1901.
Van ’t Hoff, J. H. Arrangement of Atoms in Space. Translated by A. Eiloart. Longmans, 1898.
Walker, J. Introduction to Physical Chemistry. Macmillan.
Wurtz, Ad. The Atomic Theory. Translated by E. Cleminshaw. Kegan Paul.
191
¶ Hitherto there have been few, if any, really popular works touching the historical growth of the various great branches of knowledge. The ordinary primer leaves unexploited the deep human interest which belongs to the sciences as contributing to progress and civilization, and calling into play the faculties of many of the finest minds. Something more attractive is wanted.
¶ The above need in literature has now been met. Each volume in The History of Sciences is written by an expert in the given subject, and by one who has studied the history as well as the conclusions of his own branch of science. The monographs deal briefly with the myths or fallacies which preceded the development of the given science, or include biographical data of the great discoverers. Consideration is given to the social and political conditions and to the attitudes of rulers and statesmen in furthering or in hindering the progress of the given science. The volumes record the important practical application of the given science to the arts and life of civilized mankind, and also contain a carefully-edited bibliography of the subject. Each volume contains from twelve to sixteen carefully-prepared illustrations, including portraits of celebrated discoverers, many from originals not hitherto reproduced, and explanatory views and diagrams. The series as planned should cover in outline the whole sphere of human knowledge.
¶ Science is to be viewed as a product of human endeavor and mental discipline, rather than taken in its purely objective reference to facts. The essential purpose has been to present as far as practicable the historical origins of important discoveries, also to indicate the practical utility of the sciences to human life.
G. P. PUTNAM’S SONS
New York London
Each volume is adequately illustrated, attractively printed, and substantially bound.
16mo. Each, net, 75 cents. By mail, 85 cents. 12 illustrations
History of Astronomy
By George Forbes, M.A., F.R.S., M.Inst. C.E.
Formerly Professor of Natural Philosophy, Anderson’s College, Glasgow
I thank you for the copy of Forbes’s History of Astronomy received. I have run it over, and think it very good indeed. The plan seems excellent, and I would say the same of your general plan of a series of brief histories of the various branches of science. The time appears to be ripe for such a series, and if all the contributions are as good as Prof. Forbes’s, the book will deserve a wide circulation, and will prove very useful to a large class of readers.—Extract from a letter received from Garrett P. Serviss, B. S.
History of Chemistry
By Sir Edward Thorpe, C.B., LL.D., F.R.S.
Author of “Essays in Historical Chemistry,” “Humphry Davy: Poet and Philosopher,” “Joseph Priestley,” etc.
12 illustrations. Two vols. Vol. I—circa 2000 B.C. to 1850 A.D. Vol. II—1850 A.D. to date
The author traces the evolution of intellectual thought in the progress of chemical investigation, recognizing the various points of view of the different ages, giving due credit even to the ancients. It has been necessary to curtail many parts of the History, to lay before the reader in unlimited space enough about each age to illustrate its tone and spirit, the ideals of the workers, the gradual addition of new points of view and of new means of investigation.
The History of Old Testament Criticism
By Archibald Duff
Professor of Hebrew and Old Testament Theology in the United College, Bradford
The author sets forth the critical views of the Hebrews concerning their own literature, the early Christian treatment of the Old Testament, criticism by the Jewish rabbis, and criticism from Spinoza to Astruc, and from Astruc until the present.
In Preparation
The History of Geography.
By Dr. John Scott Keltie, F.R.G.S., F.S.A., Hon. Mem. Geographical Societies of Paris, Berlin, Rome, Brussels, Amsterdam, Geneva, etc.
The History of Geology.
By Horace B. Woodward, F.R.S., F.G.S., Assistant Director of Geological Survey of England and Wales.
The History of Anthropology.
By A. C. Haddon, M.A., Sc.D., F.R.S., Lecturer in Ethnology, Cambridge and London.
The History of New Testament Criticism.
By F. C. Conybeare, M.A., late Fellow and Praelector of Univ. Coll., Oxford; Fellow of the British Academy; Doctor of Theology, honoris causa, of Giessen; Officer d’Academie.
Further volumes are in plan on the following subjects:
Mathematics and Mechanics—Molecular Physics, Heat, Light, and Electricity—Human Physiology, Embryology, and Heredity—Acoustics, Harmonics, and the Physiology of Hearing, together with Optics, Chromatics, and Physiology of Seeing—Psychology, Analytic, Comparative, and Experimental—Sociology and Economics—Ethics—Comparative Philology—Criticism, Historical Research, and Legends—Comparative Mythology and the Science of Religions—The Criticism of Ecclesiastical Institutions—Culture, Moral and Intellectual, as Reflected in Imaginative Literature and in the Fine Arts—Logic—Philosophy—Education.
G. P. PUTNAM’S SONS
New York London
Punctuation and spelling were made consistent when a predominant preference was found in this book; otherwise they were not changed.
Simple typographical errors were corrected; occasional unbalanced quotation marks retained.
Ambiguous hyphens at the ends of lines were retained; occurrences of inconsistent hyphenation have not been changed.
Index not checked for proper alphabetization or correct page references.