J. F. Illingworth
Chair of the Committee on
Advanced Degrees.
Obvious typographical errors have been silently corrected. All other spelling and punctuation remains unchanged. In particular the author uses Kavahin for what is now normally referred to as Kavain.
THESIS
presented for the degree of
MASTER OF SCIENCE
at
THE COLLEGE OF HAWAII
JUNE 1915
by
ALICE A. BALL.
The Thesis, herewith, on “The Chemical Constituents of the Active Principle of the Ava Root” by Alice A. Ball, clearly demonstrates her ability to do original work and to present her results in logical form. Approved.
J. F. Illingworth
Chair of the Committee on
Advanced Degrees.
College of Hawaii,
May 14, 1915.
OR
THE CHEMICAL CONSTITUENTS
OF THE ACTIVE PRINCIPLE
OF THE AVA ROOT.
| Page | ||
| 1. | Historical | 1 |
| 2. | Method of Extraction | 7 |
| 3. | Method of Separation of the Resins | 9 |
| 4. | Various Metallic Salts of the Resinous Acids | 12 |
| 5. | The Total Resins | 13 |
| 6. | The Barium Acid | 15 |
| 7. | Oxidation Products of the Barium Acids | 21 |
| 8. | The Iron Acids | 28 |
| 9. | Oxidation of the Iron Acids and the Free Acids | 33 |
| 10. | Alcohol Radicals | 34 |
| 11. | Methysticin and Methysticinic Acid | 36 |
| 12. | Physiological Action | 38 |
| 13. | Conclusion | 43 |
[Pg 1]
“Among the customs peculiar to the inhabitants of the South Pacific Islands, perhaps the most noted is that of the preparation and drinking of a narcotic beverage called ava, kava, or yakona. Much of its notoriety arises from the repulsive way in which it is sometimes made. Aside from this, it is characteristic of a certain oceanic area, and seems to be as strikingly limited to this area as is the stick-and-groove method of making fire. The custom, is not confined to one ethnic stock, many notices in literature showing that both Papuans and Polynesians practise it. In many of the islands the Liquor is concocted by chewing the root of the Macropiper methysticum, or long pepper, ejecting the comminuted mass into a bowl, adding water, straining out the pulp, and drinking the fluid. In other localities it is made by simply grating the root and adding water.
“The plant from which kava is made is a shrub of the natural order Piperaceae. It is about six feet high with stems ranging from an inch to an inch and a half in thickness; the leaves are cordate and from four to eight inches long. This family is the source of the pepper of commerce and contains several species that are of medicinal and commercial importance.
[Pg 2]
In making kava, the root and base of the stem is used. The roots usually weigh from two to four pounds, though sometimes as much as 22 pounds. Several varieties are distinguished by the natives; for instance, in Tahiti there is a yellow variety called Marea; another, which becomes pink on exposure to the air, is called avini-ute.
“Chewed when freshly gathered, the root first tastes sweet and aromatic, then bitter, acrid and pungent. It provokes abundant secretion of saliva and in a few seconds occasions a sensation of burning on the tongue. The root contains about fifty percent of starch, a little pale-yellow essential oil, two percent of an acrid resin, and one percent of the neutral crystalline principle methysticin, called kavahin. To the latter principle we must attribute the toxic qualities of the kava preparation. The resin and the kavahin are insoluble in water, but are soluble in saliva and the gastric juices.
“In Samoa, the ava root is grated or chewed, then soaked, the woody pulp strained off, and the fluid drunk. The root is used either dry or green. The flavor of the liquid is at first like that of soapsuds, but immediately afterward a pleasant aromatic taste is imparted, faintly bitter, as in quinine. In Samoa, ava drinking is the accompaniment of all meetings of the men.
[Pg 3]
“Kava is at first stimulating, but the effect of an excess resembles that of opium, producing a drowsy drunkenness, lasting for two hours. The inebriate is usually peaceable, but sometimes is irritated by noises, which is attributed by natives to the use of kava grown in moist ground. The results of excess are skin disease, emaciation, and general decrepitude. The peculiar whiteness of the skin caused by kava drinking is said to be sought after in some islands as a sign that its possessor is wealthy enough to devote his time to its acquirement.
“There is some misapprehension in regard to whether the liquid undergoes fermentation before it is consumed, but it is positively known that there can be no fermentation, for the liquor is drunk immediately after the addition of water to the macerated root. Kava that is prepared by chewing is said to be more palatable, which is perhaps due to the conversion of the starch into a fermentable substance by the ptyalin of the saliva.”[1]
[1]—By Walter Hough—Reprinted from Smithsonian Miscellaneous Collections—No. 1472—August 1904. “Kava Drinking as Practised by the Papuans and Polynesians.”
“In 1779 Captain King, R. N., who followed Captain Cook to the Islands, describes the case of a priest as follows ‘a little old man of an emaciated figure, his eyes exceedingly[Pg 4] sore and red, and his body covered with a white leprous scurf, the effects of an immoderate use of ava.’ He also says, ‘The chiefs suffer dreadful effects from the immoderate use of ava. Those who are most affected by it had their bodies covered with a white scurf, their eyes red and inflamed, their limbs emaciated, their whole frame trembling and accompanied with a disability to raise the head.’”[2]
[2] “Leprosy Prize Essays,” 2nd series by Thompson and Cantile, 1897.
F. A. Griel, makes the following statement in a foot note. “The mixture is a subnarcotic, and if drunk by European sailors produces highly nauseous effects. If frequently taken a dry burning heat is produced all over the body, the eyes become red, skin peels off in flakes and then degenerates into leprous ulcers or the whole body becomes emaciated and wastes away.”[3]
[3] Miquel, Systema Piperacearum.
Numerous attempts have been made to isolate the active chemical constituent or constituents. As early as 1844 Morson discovered an active principal Kawine. This is a greenish-yellow, strongly aromatic and acrid resin. This was again studied by Cuzant in 1860.
[Pg 5]
Gobley isolated from kava root a crystalline principle (analogous to piperin), methysticin, or kavahin, which is without odor and taste and is probably inert.[4] In 1886 Lewin separated the resin into two resins, of which the Beta resin is greasy and of a reddish-brown color, appearing in mass almost black. This is less active than the alpha resin which is yellowish brown, has the characteristic odor of the drug, is freely soluble in alcohol, and placed upon the tongue produces a burning sensation followed by local anaesthesia.[5]
[4] J. P. C. Jan. 1860.
[5] A. J. P. 1886, 450.
A volatile oil has been found in the root.[6]
[6] J. P. C. March 1862.
Lavialle claimed to have obtained an alkaloid, Kavaine.[7]
[7] L’Union Pharm. Jan. 1889.
The following statement was found in “Watts Chemical Dictionary”, “Kawain—a crystalline resin occurring along with methysticin in kawa-kawa, It is not a glucoside. On oxidation it yields benzoic acid.[8]
[8] Gobley, J. Ph. (3) 37, 19.
[Pg 6]
The following statement appears in the Encyclopedia Britannica. “There appears to be little doubt that the active principle in this beverage is a poison of an alkaloidal nature. It seems likely that this substance is not present as such (i.e. as a free alkaloid) in the plant, but that it exists in the form of a glucoside, and that by the process of chewing, this glucoside is split up by one of the ferments in the saliva and the free alkaloid and sugar is formed”.
Arthur Bossingham[9] communicates the results of a chemical examination of Kava-kava. Besides the crystalline body, methysticin, which has already been described by others, he was able to isolate and identify three resins, one soluble in 5% solution of potassium carbonate, the second insoluble in this, but soluble in 5% solution of caustic potash, while the third was insoluble in both of these alkaline solvents, The ash amounts to 2.495% of the air dried root, and contained besides mere traces of Fe, Mn, mainly Calcium, Sodium and Potassium.[10]
[Pg 7]
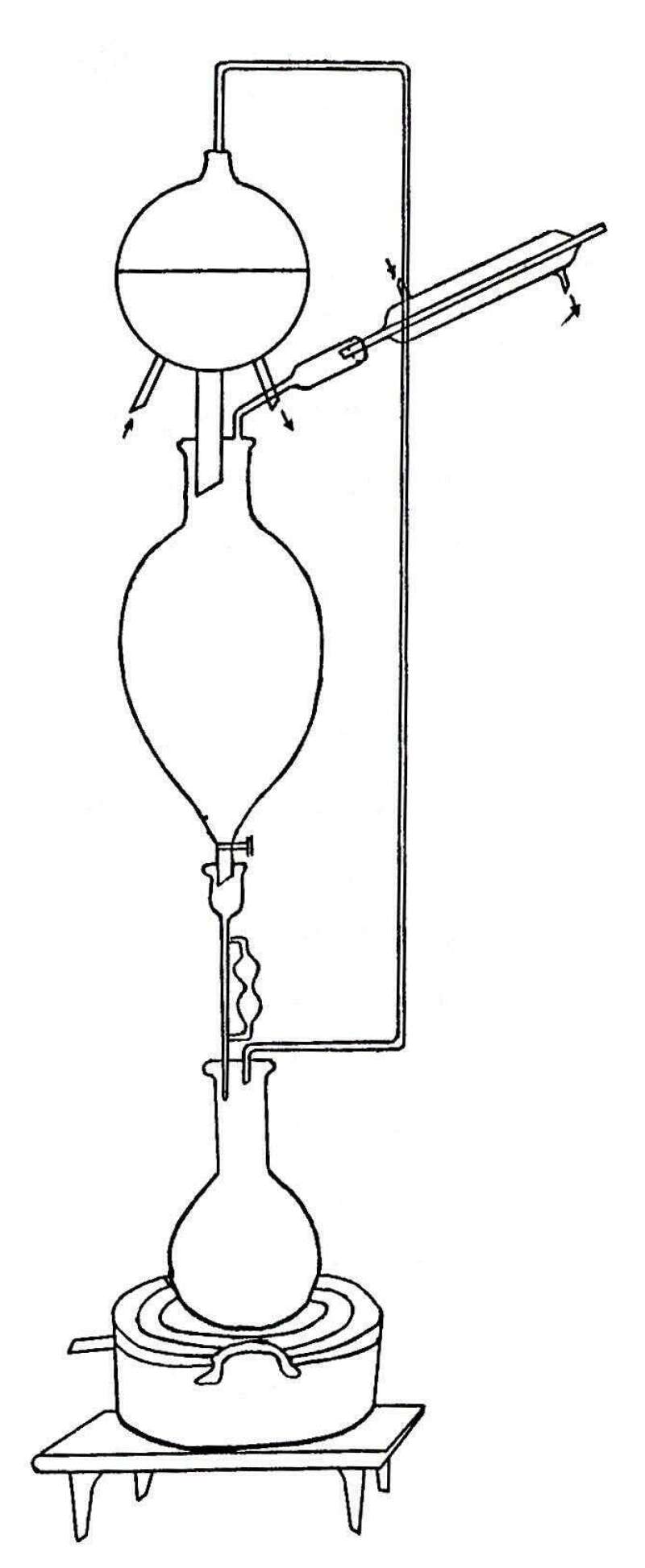
The fresh rhizome was chopped up and then ground up by means of a meat chopper. In the preliminary work the material was dried in a vacuum oven at a temperature not exceeding forty degrees Centigrade. In the later work the material was rapidly dried in the sun. This operation required about two days time. After being thoroughly dried, the material was finely powdered, and then extracted with ether. The following continuous extraction apparatus was used. Due to the extreme rapidity with which the ether evaporated, it was necessary to surround the coils with ice so as to keep the condensers cold.
[Pg 8]
[Pg 9]
After the removal of the crystalline methysticin from the extract, the following method of separation of the resinous products was used.
The free acids were removed by shaking the total resins successively with solutions of one percent ammonium carbonate, one percent sodium carbonate and one percent sodium hydroxide. The acids thus removed were recovered by treating the alkaline solution with dilute hydrochloric acid and shaking out with ether. The three acids were not markedly different in their physical properties. They possessed the characteristic odor of the crude drug, were viscous, brown in color and did not solidify at minus ten degrees Centigrade. These free acids constitute about five percent of the total resins.
The remainder of the extract was treated with alcoholic potassium hydroxide and saponified by heating the solution to about eighty degrees for fifteen minutes. After the alcohol was removed by distillation a small quantity of water was added. This resulting solution was then extracted with ether to remove the alcohol radical or radicals and the unsaponifiable material from the saponified product. The ether extract was saved to be used in the work on the[Pg 10] alcohol radicals. The aqueous portion was evaporated to a semi-solid mass. Carbon dioxide was passed over this mass for about fifteen minutes to change any excess of potassium hydroxide to potassium carbonate. Alcohol (95%) was then added to precipitate the carbonate. The carbonate was then filtered off and water added to the filtrate.
To the clear filtrate a solution of barium chloride was added. Immediately a dense thick cream yellow precipitate formed. Barium chloride was added in excess. The barium salt was filtered off by means of a suction pump and the precipitate washed with water. The filtrate was treated with a few drops more of barium chloride so as to be sure that an excess had been added, which was proven if no further precipitate formed. To this clear filtrate an excess of a solution of ferric chloride was added. Immediately a thick heavy precipitate of the iron salt came down, which was filtered off by means of a suction pump and the precipitate washed with water.
The barium precipitate was treated with dilute hydrochloric acid and heated to boiling to decompose the barium resinate, and to liberate the free resin acid. The liberated acid was a brown resin of a thin consistency and had a characteristic odor. This acid was removed by[Pg 11] shaking out with ether. The ethereal solution was dried with anhydrous sodium sulphate, filtered, and the ether removed by distillation. The last traces of ether were removed by heating the resin acids in a vacuum oven at sixty five degrees Centigrade. This resinous acid is the acid spoken hereafter in this paper as the BARIUM ACID.
The iron precipitate was treated with dilute sulphuric acid (ferric chloride is soluble in ether and ferric sulphate is not) and heated to boiling to decompose the iron resinate. The acid when liberated had a strong characteristic aromatic odor. It was extracted with ether in the same manner as the barium acid. This resinous acid is the acid spoken of in this paper as the IRON ACID.
The resinous extract left after the removal of the crystalline methysticin is spoken of as the TOTAL RESINS.
The resin acids removed by the preliminary shaking out with the various aqueous alkalies are spoken of as the TOTAL FREE ACIDS.
[Pg 12]
After obtaining the potassium salts of the acids and having freed the same from the excess of the potassium hydroxide and potassium carbonate, the possibilities of the formation of different metallic salts were tried.
The soluble salts of the following metals yielded precipitates:—
The following metallic salts gave a complete precipitation:—
Since the barium and iron (ferric) salts gave the best means of separation, they were used to separate the resins.
[Pg 13]
The total resins were brown in color, thick syrupy consistency and possessed the characteristic odor of the drug.
A molecular weight determination was made in the same manner as with the Barium acids, and the following data obtained.
| Wt. of pipette before | 18.7660 |
| Wt. of pipette after | 14.7160 |
| Wt. of resin used | 4.0500 |
| Temperature before | 3.52 |
| Temperature after | 3.71 |
| Change in temperature | .18 |
| Volume | 37 cc. |
| Constant for solvent | 3280 |
| Approximate molecular weight | 2000 |
Combustions were made and the following data obtained.
| Wt. of boat | 3.4722 | 3.4720 |
| Wt. of boat and resin | 3.6355 | 3.6220 |
| Wt. of resin | .1633 | .1500[Pg 14] |
| Sulphuric acid tube | 79.1051 | 79.3865 |
| Tube plus water | 79.2073 | 79.4770 |
| Water | .1022 | .0905 |
| Hydrogen equivalent | .01136 | .01006 |
| KOH bulb | 49.4316 | 50.8720 |
| Bulb plus carbon dioxide | 49.8350 | 51.2430 |
| Carbon dioxide | .4034 | .3710 |
| Carbon equivalent | .12102 | .1010 |
| Percent Carbon | 67.31% | 67.3 |
| Percent Hydrogen | 6.9 % | 6.7 |
| Percent Oxygen | 26.8 % | 27.0 |
The total resins when placed on the end of the tongue produced a marked stinging sensation followed by a local anaesthesia. After the first stinging was produced the sensation was rather pleasant. The local anaesthesia persisted a long time, giving a sensation much the same as that produced by cocaine. The barium and iron acids also produced this local anaesthesia, but the initial stinging sensation was much more pronounced, which was probably due to the acid nature of the substance.
[Pg 15]
The resinous material used and spoken of as the “Barium Acid” is the material prepared and so named as given under the “Method of Separation”.
PHYSICAL PROPERTIES:—Dark reddish brown in color, syrupy in consistency and has a characteristic odor; heavier than water; soluble in benzol, ether, alcohol and acetone, but insoluble in petroleum ether and water.
This resin constitutes about sixty percent of the total ester resins; i.e. the resins left after the free acids have been removed with aqueous potassium hydroxide.
An analysis of the barium salt obtained by precipitation from the potassium soap was made and the following data obtained:—
| Wt. of substance used | 1.1715 | .5860 |
| Wt. of barium sulphate | .4430 | .2205 |
| Barium equivalent | .2606 | .1298 |
| Percent barium | 22.2% | 22.1% |
The following method was used in making the above analysis. The weighed material was ignited in a platinum crucible by gently heating until the combustible gases formed were given off. The crucible was then more[Pg 16] strongly heated to completely burn off the carbonaceous material left. The residue was extracted with nitric acid and the barium precipitated as the sulphate with dilute sulphuric acid, and the weight of the barium sulphate determined.
Expressed as the ACID NUMBER, or the number of milligrams of potassium hydroxide required to neutralize the free acids in one gram of the substance, the following data was obtained:—
| Barium equivalent | .2606 | .1298 |
| Barium expressed as KOH equiv. | .2131 | .1062 |
| Wt. of material used | 1.1715 | .5860 |
| Milligrams of KOH per gram | 181.9 | 181.3 |
| ACID NUMBER | 181.9 | 181.3 |
The following gives the ACID NUMBER obtained by direct titration of the barium acid; in “A”, barium hydroxide was used and in “B”, sodium hydroxide was used.
A small quantity of the material was dissolved in a sufficient quantity of neutralized alcohol to give a liquid of a light yellow color, Phenolphthalein was used as the indicator, and the alkali was added until a red color was produced.
[Pg 17]
| “A” | “B” | |
| Wt. of substance | .6160 | .2345 |
| Cc. of alkalie | 4.54 | 1.75 |
| KOH equiv. per gram | 41.40 | 41.90 |
| Acid number | 41.40 | 41.90 |
On evaporating a portion of the alcohol from the material left after titrating with the sodium hydroxide, and adding water to obtain an aqueous solution of the sodium salt, an emulsion was formed, and on standing globules of the free resinous acid separated. From the data thus obtained, it can readily be seen that the acid or acids which constitute the BARIUM ACID must have a number of carboxyl groups and form a different series of salts by precipitation than by direct titration. The salt or salts formed by direct titration, although neutral to phenolthalein may be acid in structure. This is further shown by the fact that the potassium salts produced by direct titration are readily hydrolyzed. The acid number obtained by the precipitation of the barium salt may be called the COMBINING VALUE, and the acid number obtained by titration the TITRATION VALUE.
A number of molecular weight determinations were[Pg 18] made on the free barium acid. McCoy’s Boiling Point Apparatus was used and Merk’s benzol (free from thiophene) was used as the solvent. A weighing pipette with a bulb was used to introduce the material, the bulb being weighed before the material was introduced into the apparatus, and afterwards, the difference being the weight of the material used.
| Wt. of pipette (before) | 16.3670 | 14.8895 |
| Wt. of pipette (after) | 14.8895 | 12.4610 |
| Wt. of material used | 1.5225 | 2.4285 |
| Original temperature | 3.53 | 3.53 |
| Final temperature | 3.60 | 3.62-3 |
| Change in temperature | .07 | .09 |
| Volume of solution | 37 cc | 43.5 cc |
| Constant for solvent | 3280 | 3280 |
| Approx. Molecular Wt. | 2000 | 2100 or 1800 |
Combustions were made using the barium acids. By qualitative tests it was found that the acids contained only carbon, hydrogen and oxygen. The following gives the results of the combustions.
| Wt. of boat | 2.8402 | 2.8402 |
| Wt. of boat and resin | 3.0500 | 3.0250 |
| Wt. of resin | .2098 | .1848[Pg 19] |
| Wt. of H2SO4 tube | 78.0250 | 78.1415 |
| Wt. of tube plus water | 78.1520 | 78.2550 |
| Wt. of water | .1270 | .1135 |
| Hydrogen equiv. | .0143 | .0126 |
| Wt. of KOH bulb | 48.8140 | 51.7095 |
| Wt. of bulb plus CO2 | 49.3800 | 52.2085 |
| Wt. of CO2 | .5660 | .4990 |
| Carbon equiv. | .1543 | .1361 |
| Percent Hydrogen | 78.5% | 73.6% |
| Percent Carbon | 6.8% | 6.8% |
| Percent Oxygen | 19.7% | 19.6% |
An attempt was made to make the potassium salts of the barium acids by saponification with alcoholic potash. A small quantity of the acid was treated with an excess of ten percent alcoholic potash and heated to eighty degrees Centigrade to complete the saponification. Instead of the formation of the potassium salts, a thick dark brown solid, gummy mass separated. On cooling it solidified to a brittle solid which had all the physical properties of a true resin. This solid is soluble in ether, chloroform and benzol,[Pg 20] slightly soluble in alcohol and insoluble in petroleum ether and water. It burns without the formation of an ash. Evidently, this brittle material is a condensation product of the original barium acid.
[Pg 21]
A small amount of the Barium acids was sealed with concentrated nitric acid in a hard glass tube and heated in a bomb furnace for an hour and a half at 115 to 120 degrees Centigrade. On cooling a yellow solid separated. Qualitative tests showed that this oxidation product contained no nitrogen, combined with sodium hydroxide readily, is soluble in hot water, slightly soluble in cold water, easily soluble in ether, alcohol and benzol and slightly soluble in carbon tetrachloride. It decolorizes alkaline permanganate but does not decolorize bromine water.
That there are a number of intermediate products formed and that nitration also takes place during the formation of these intermediate products is shown by the following. A small quantity of the acids were placed in a test tube and covered with concentrated nitric acid. This was suspended in an H2SO4 bath and a thermometer inserted so as to observe the temperature. On being gently warmed the nitric acid and the resin began to react with a rapid evolution of carbon dioxide and oxides of nitrogen. As soon as the reaction had modified and before the temperature rose above one hundred degrees Centigrade, a small amount was removed and added to water. Some of the solid acid was formed[Pg 22] and also a number of globules of oil and there was a strong persistent odor of nitrobenzene. The original test tube was heated to about one hundred and twenty degrees centigrade and a small portion again removed. There was more of the solid material formed and the odor was similar to vanillin or coumarin or cinnamic aldehyde. The test tube was again tested when the temperature had reached one hundred and thirty five degrees Centigrade. There was no aromatic odor and a large amount of the solid formed. When viewed under the microscope the substance had the appearance of curled threads.
When the barium acids were treated with the standard nitrating mixture, a solid was obtained which showed the presence of nitrogen when the standard test was applied.
After the preliminary tests were made, the following method of preparation was used. Two or three grams of the barium acids were introduced into a hard glass tube of about thirty centimeters in length and fifteen or twenty cubic centimeters of concentrated nitric acid added. The reaction which is very vigorous at first was regulated by keeping the tube under running water. After this vigorous action was over the tube was placed in a sulphuric acid bath, and the temperature gradually increased until it had[Pg 23] reached one hundred and twenty five degrees Centigrade, at which temperature it was kept for about five hours. It was necessary to add a small quantities of nitric acid from time to time to make up the loss by evaporation. When the oxidation was completed the product was poured into water and then heated to boiling. The resulting solution was filtered and the filtrate allowed to cool. On standing a quantity of a pale yellow substance separated. The following data gives the percent yield of this oxidation product.
| Wt. of container | 6.7220 |
| Wt. of container and substance | 7.5920 |
| Wt. of substance | .8700 |
| Wt. of oxidation product | .1240 |
| Percent yield | 14% |
The oxidation product was dried by placing it in a vacuum over sulphuric acid for several days. The neutralization equivalent of this crude oxidation product was 157.
| Wt. of substance used | .0502 |
| Number Cc. of NaOH N/10 | 3.2 cc. |
| Neutralization equivalent | 157 |
The oxidation product was heated on a watch crystal and the sublimate allowed to collect on a funnel. The first sublimate gave a melting point of 109 degrees Centigrade.
[Pg 24]
Combustions were made on this sublimate with the following results.
| (1) | C | 66% |
| H | 4.7% | |
| (2) | C | 65.7% |
| H | 4.8% | |
| (3) | C | 65.15% |
| H | 4.8% | |
| (4) | C | 65.8% |
| H | 4.85% |
The neutralization equivalent was obtained by titrating an alcoholic solution of the sublimate with standard sodium hydroxide. The following results were obtained on two different lots of the sublimed oxidation product.
| Wt. of substance | .09 | .1006 |
| Cc. of alkali N/10 | 7 cc | 7.9cc |
| Neutralization equiv. | 128.6 | 127.3 |
Using a third sample the neutralization equivalent was obtained from the analysis of the silver salt. The silver salt was formed by adding silver nitrate solution to a carefully neutralized solution of the sublimate. The insoluble silver salt was filtered off, washed with water to re[Pg 25]move the excess of silver nitrate, and dried in a vacuum over sulphuric acid for several days. A weighed quantity of the silver salt was ignited in a platinum crucible and the residue of metallic silver was weighed. The following data were obtained using material from the same sample for each analysis.
| Wt. of dish | 12.8825 | 12.8826 |
| Wt. of dish and substance | 13.0060 | 13.1310 |
| Wt. of substance | .1235 | .2484 |
| Wt. of dish and silver | 12.9400 | 12.9980 |
| Wt. of silver | .0575 | .1154 |
| Neutralization equiv. | 125 | 125.3 |
Using the same sample, a neutralization equivalent was obtained by titration with standard NaOH.
| Wt. of substance | .0912 |
| N/10 NaOH | 7.25cc |
| Neutralization equiv. | 125.8 |
The above data shows that the sublimate is a mixture. No empirical formula can be calculated from the combustions, and different samples give different neutralization equivalents although the duplicate determinations on the same sample showed good agreement thus demonstrating[Pg 26] the reliability of the methods.
By fractional sublimation it was possible to obtain fractions with different melting points. The first sublimate melted sharply at 109 degrees. From the last fraction it was possible to separate some crystals that melt at 200 degrees Centigrade. These might possibly be p-acetyl-benzoic acid, as its properties of solubility, crystalline form, its melting point and power of sublimation agree with those of p-acetyl-benzoic acid.
Those crystals that appeared identical with benzoic acid were placed in a melting point tube, and some known benzoic acid (from toluol) was placed in another tube. These two tubes were placed in the same sulphuric acid container and their melting points taken at the same time. They melted at the same temperature.
The sublimate had a very pleasant aromatic odor resembling benzoin. It gave no coloration with ferric chloride, thus eliminating a large group of aromatic compounds. Some of the crystals were found to be identical with benzoic acid when examined under the microscope. The characteristic odor of methyl benzoate was produced when a small quantity of the crystals were heated with methyl alcohol and concentrated sulphuric acid. On treating some of the carefully[Pg 27] neutralized product with ferric chloride solution, a flesh colored precipitate was formed. It agreed closely in its analysis with the precipitate formed with known benzoic acid.
The filtrate left after the removal of the iron precipitate was acidified and extracted with ether, and the ether removed by evaporation. The resulting substance decolorized alkaline permanganate solution, but did not decolorize bromine water. When the leaflet needles that melt at 200 degrees were mechanically removed from the original sublimate, the substance left after precipitating with ferric chloride melted at 109 degrees. When these crystals were not removed, the melting point of this material was not definite, but was over a range of five degrees, from 110 to 115 degrees Centigrade.
The oxidation product contains at least three distinct substances, benzoic acid, a substance melting at 200 degrees and—which is probably p-acetyl benzoic acid and a third substance melting at 110 degrees.
[Pg 28]
The resinous material used and spoken of as the IRON ACIDS is the material prepared and so named under the “Method of Separation”.
PHYSICAL PROPERTIES:—Transparent and reddish brown in color, oily in consistency and has a characteristic tea like odor, heavier than water, freely soluble in benzol, ether, alcohol and acetone, but insoluble in petroleum ether and water.
This resin constitutes about eighteen percent of the total ester resins.
By qualitative tests it was shown that the acids contained only carbon, hydrogen and oxygen. Combustions made on the iron acids gave the following results.
| Wt. of boat | 2.6950 | 2.6950 |
| Wt. of boat and substance | 2.8470 | 2.8495 |
| Wt. of substance | .1520 | .1545 |
| KOH bulb | 50.9620 | 51.0805 |
| Bulb and CO2 | 51.3370 | 51.4610 |
| Wt. of CO2 | .3750 | .3805 |
| Carbon equivalent | .1023 | .10376[Pg 29] |
| Sulphuric acid tube | 76.2448 | 76.3450 |
| Tube and water | 76.3400 | 76.4453 |
| Wt. of water | .0952 | .1003 |
| Hydrogen equiv. | .0106 | .01114 |
| Percent Carbon | 67.3% | 67.2% |
| Percent hydrogen | 7.0% | 7.2% |
| Percent Oxygen | 25.7% | 25.6% |
An analysis of the iron salt obtained by precipitation from the potassium soap gave the following data.
| Wt. of substance used | .2955 | .3387 |
| Wt. of FeSO4 | .0445 | .0509 |
| Ferric equiv. | .03208 | .03676 |
| Percent Iron | 10.85% | 10.85% |
The following method was used in making the above analysis. The weighed material was ignited in a platinum crucible by gently heating until the combustible gases formed were given off. The crucible was then strongly heated until the carbonaceous material was completely burned off. The residue was weighed and the percentage of iron determined.
Expressed as the ACID NUMBER, or the number of milligrams of KOH required to neutralize the free acids in[Pg 30] one gram of the substance, the following data was obtained.
| Ferric equivalent | .03208 | .03676 |
| Fe expressed as KOH equiv. | .0965 | .1107 |
| Wt. of material used | .2955 | .3387 |
| Mg. of KOH per gram | 326.6 | 326.7 |
| Acid number | 326.6 | 326.7 |
The following gives the ACID NUMBER obtained by direct titration of the Iron acid. The method is the same as that used in getting the titration value of the Barium acid.
| Wt. of substance | .2450 | .2472 |
| Cc of alkalie | 2.2 | 2.3 |
| KOH equiv. | .01232 | .01288 |
| KOH equiv. per gram | 53.87 | 52. |
[Pg 31]
An attempt was made to saponify some of the iron acid, but it was impossible. The alcohol was partially distilled off, and the acid freed by making the mass acid with sulphuric acid, and shaking out with ether. The ether was distilled off, but the remaining acid had different physical properties from the acid with which the experiment was started. It was lighter in color, and solidified at zero degrees. At room temperature it was almost solid. On ignition it left no ash. This probably is a polymerization product of the original acid.
Since many organic acids whose salts cannot be prepared by the ordinary methods can be prepared by passing dry ammonia gas through a solution of the acid in anhydrous ether, this method was tried with the iron acid. The iron acid was dissolved in anhydrous ether, and the dry ammonia gas was bubbled through this ether solution. At first no change was noted, but after several minutes there was a flocculent thready precipitate formed which was light brown in color. The experiment was repeated. At first the precipitate was a very light brown, but after forming it quickly darkened. After standing a few hours the flocculent precipitate changed to a sticky brown mass. This same change was produced immediately if the precipitate was exposed to the air. The resulting mass had no odor of ammonia.
[Pg 32]
The flocculent precipitate formed at first was probably the ammonium salt of the iron acid, which like most ammonium salts, it was precipitated due to its insolubility in ether. Due to the ease of hydrolysis this salt immediately decomposed to the acid and ammonia when traces of moisture were present.
[Pg 33]
When the iron acids were oxidized in the same manner as the barium acids the amount of the oxidation product formed was about one fourth of that produced with an equal amount of the barium acids. The time necessary to completely oxidize the iron acids was much less then required for the barium acids. On sublimation the iron acid gave a product melting at 110 degrees Centigrade and is probably identical with the one formed from the barium acids. The iron acid also yielded a sublimate melting at 208 degrees C.
The free acids when oxidized in a like manner gave as one of the products a low melting crystalline compound that contained nitrogen.
The iron acid although related to the barium acid as shown by the formation of a common oxidation product is different in structure as shown by the difference in the amount of the oxidation product formed and the time to complete the oxidation.
[Pg 34]
The material that was shaken out with ether after the saponification consists of the unsaponifiable material and the alcohol radicals of the acids produced by saponification.
On allowing the ether to evaporate from this material, feathery needles separated. These were removed by means of a suction pump and recrystallized from hot acetone. The melting point of this product was 122-125 degrees Centigrade. The precipitate was dissolved in benzol and allowed to slowly crystallize. It formed long prismatic needles with melting point of 130 degrees Centigrade. When heated with concentrated sulphuric acid, a brown green fluorescent solution was produced.
The remaining material was steam distilled, and the distillate extracted with ether to remove the oil. The ether was dried with anhydrous sodium carbonate, the solution filtered and the ether removed by distillation. The oil that remained was light yellow, specific gravity less than that of water, and possessed a very characteristic odor resembling that of musk. The material left after the steam distillation was cooled and shaken out with ether. The ethereal solution[Pg 35] was dried with anhydrous sodium sulphate and the ether removed by distillation. The resulting mass was a dark brown resin without any characteristic odor, solidifying at zero degrees.
The crystalline product with a melting point of 130 degrees and the essential oil are evidently the alcohols formed through saponification of the resin esters. The crystalline product could be readily separated from the resinous material because of its slight solubility in acetone. Since this product was not precipitated when the unsaponified resin esters were treated with acetone and because it did not make its appearance until after the process of saponification, it is quite probable it is a product of saponification. The essential oil can be detected in extremely small amounts due to its penetrating characteristic odor. Before the process of saponification it could not be detected, but as soon as saponification took place the odor was very marked. From this it is quite evident that this oil is a product of saponification.
The dark brown resinous mass that remained may be either an alcoholic resin formed through the hydrolysis of the ester resins or it may be a resin belonging to the class known as resenes[11] which resist saponification.
[11] Com. Organic Analysis. Allen Vol. II, 146.
[Pg 36]
The crystalline product obtained from the ether extract was recrystallized several times from absolute alcohol to remove all traces of any resinous material. It had a melting point of 122-123. It therefore was not pure methysticin, which has a melting point of 138-139 degrees. On examining this product under the microscope it was found to contain two distinct forms of crystals, long needles and prismatic plates. Some free methysticinic acid, melting point 180 was examined under the microscope and found to consist entirely of prismatic plates. Therefore the original crystalline body contains some methysticinic acid.
Some of the original precipitate was crystallized once from absolute alcohol and thoroughly dried. This was used for the following data to determine the percent of free methysticinic acid in the crystalline product. A weighed amount of the crystalline product was dissolved in carefully neutralized alcohol, and titrated with tenth normal NaOH, using phenolthalein as the indicator.
| Wt. of substance used | .2590 |
| NaOH N/10 | .5 cc |
| % methysticinic acid. |
[Pg 37]
The potassium salts of the various resin acids formed by the saponification of the ester resins should yield a resinous mass syrupy or oily in consistency when acidified, if it consists entirely of the two groups of acids spoken of as the iron acids and the barium acids. Also the weight of the barium acids plus the weight of the iron acids should nearly equal the weight of the ester resins, if these are the only two acids, because the alcohol radicals constitute not more than two percent of the ester resins. But when the total potassium salts were acidified the product formed contained a crystalline substance in addition to the resinous acids. This crystalline substance was separated from the resinous acids by means of their difference in solubility in ether. On recrystallization from absolute alcohol, it had a melting point of 179 degrees Centigrade. It is therefore methysticinic acid. This acid constitutes about twenty percent of the total acids from the ester resins because the barium and iron acids constitute only about seventy five percent.
This methysticinic acid cannot be entirely formed by hydrolysis of methysticin which was incompletely removed from the resinous material. The methysticinic acid existed either combined with some other alcohol than methyl alcohol or was combined with one of the resin alcohol radicals.
[Pg 38]
Due to the impossibility of preparing the alkali salts of the barium and iron acids so as to be positive that the resulting preparations completely represented the barium and iron acids, and due to the insolubility of the acids and the total resins in solvents from which they would not again be precipitated when introduced into the circulation, it was found necessary to make an emulsion. The emulsifying agent used was acacia (gum arabic). It was possible by careful preparation to make a permanent emulsion that was miscible with water in all proportions to form a homogeneous mixture. The animals used were rabbits of about six pounds weight. The injections were made by Dr. Geo. McCoy, director of the Leprosy Investigation Station at Kalihi. All the physiological experiments were made at the bacteriological laboratory at the Leprosy Investigation Station.
One cubic centimeter of the emulsion of the total resinous extract (strength 1 in 5) was injected into the ear of a rabbit; the animal died immediately. Another rabbit was injected in the same manner with one half cubic centimeter[Pg 39] of the same preparation. The animal immediately stretched out and became rigid, or appeared to be paralyzed, but after several minutes these symptoms lessened and after about five minutes the rabbit appeared normal as far as activity was concerned. As soon as the rabbit had recovered a second injection of one quarter of a cubic centimeter of the same preparation was injected in the same manner into the same animal. The same symptoms were produced and with equal intensity, but ten minutes passed before the animal again became conscious. For several minutes after recovery the animal appeared somewhat drowsy and stupid but it soon regained its former activity.
One half cubic centimeter of the iron acid emulsion (strength 1 in 7) was injected into the ear of a rabbit. Immediate paralysis and apparent anaesthesia set in lasting very pronouncedly for eight minutes. The rabbit’s head was drawn backwards and its legs stiffened giving symptoms similar to strychnine poisoning but they did not persist. When a second injection of one quarter cubic centimeter was given to the same animal, the same symptoms were produced, the animal remaining under the influence of the injection for about fifteen minutes.
[Pg 40]
The iron acid injection was repeated on another rabbit with the same pronounced symptoms of strychnine poisoning but the effect lasted only about ten minutes.
One half cubic centimeter of the barium acid emulsion (strength 1 in 7) was injected into the ear vein of a rabbit. The animal uttered several loud cries and after moving several feet it became spastic and went into a sort of a stupor, beginning to come out of it after ten minutes. As soon as the animal recovered, a second injection of one quarter cubic centimeter was made. The animal again uttered loud cries and then it went into a stupor, but it was not spastic. This lasted approximately ten minutes. The animal did not completely recover until about twenty minutes.
Thinking that the action might be largely mechanical and that the symptoms produced were from the emulsion itself and not from the effect of the material that was emulsified, an emulsion of olive oil was used in a like manner. This olive oil emulsion was made in approximately the same consistency as the resinous emulsions, and one cubic centimeter was injected into the ear vein of a rabbit. No visible effects followed this injection during the one hour’s time the rabbit was under observation.
[Pg 41]
One cubic centimeter intraperitoneal injections of the total resinous extract emulsion and the same amount of the iron acid emulsion were made into rabbits. During the three hours the animals were under observation no symptoms were produced. This may have been due to the extreme slowness of absorption as compared with the rate of elimination from the circulation.
Dr. J. F. Illingworth states as the result of his observations while in the Fiji Islands that the kava beverage even when taken in large amounts, does not apparently affect the brain to an extent as to cause the drinker to appear as if under the influence of alcohol, but he appears as if the muscles from the hips downward are paralyzed. These symptoms last for about half an hour, at the end of which time the person is perfectly able to walk home.
In general the active constituent of any drug produces a more pronounced but more fugitive effect than the crude drug itself, and one might therefore expect more violent reactions from the isolated active constituents of the Ava then from the crude infusion. Judging from the negative results obtained from the injection of the olive oil emulsion, it is probable that the physiological effects[Pg 42] described above following the intravenous injections of the various preparations were not mechanical but must be ascribed to the action of the constituents of the Ava.
[Pg 43]
The crystalline product consists, largely of methysticin, the methyl ester of methysticinic acid. In addition to this there is about five percent of the free methysticinic acid present.
The resinous product consists of about five percent of free resinous acids, which acids can be separated into three different acids or groups of acids according to their solubility in the different alkalies. The remaining resinous product is composed of ester resins. On hydrolysis these esters yield three distinct acids, two resinous acids and methysticinic acid. The two resinous acids are distinctly different, both in physical properties and in chemical properties. They can be sharply separated by means of the difference in solubility of their barium and iron salts. These acids may be separate individual substances, or a group of related substances. The barium and iron acids although different chemically, have some groups in common as shown by the formation of a common oxidation product. There are at least three alcohol radicals formed through hydrolysis, a resinous alcohol radical, a crystalline substance with a melting point of 130 degrees, and a volatile oil.
[Pg 44]
The physiological action of the Ava is due to the ester resins. The two resinous acids formed through the hydrolysis of these esters seem to be the two active constituents of the resin ester.
From the above outlined work it appears that the ava root does not contain any alkaloidal substance, as none of the constituents were found to contain nitrogen.
That the aqueous infusion used by the ava drinkers contains the same constituents as are extracted by the ether is shown by the following. An infusion of the ava was made by allowing some of the powdered drug to remain in contact with water for about twelve hours. The infusion was then filtered and the filtrate extracted with ether. A resinous mass remained which from all appearances was identical with the resins obtained from the ether extract. It also gave the same action when placed on the tongue as was produced by the resins from the ether extract.