Figure. 1.
Apparatus for Crushing Mineral Fertilizers.
A MANUAL FOR THE ESTIMATION OF
SOILS,
FERTILIZERS, AND
AGRICULTURAL PRODUCTS.
FOR THE USE OF ANALYSTS, TEACHERS, AND STUDENTS
OF AGRICULTURAL CHEMISTRY.
VOLUME II.
FERTILIZERS.
BY HARVEY W. WILEY,
Chemist of the U. S. Department of Agriculture.
EASTON, PA.,
Chemical Publishing Co.,
1895.
COPYRIGHT, 1895,
By Harvey W. Wiley.
In this volume an attempt has been made to treat the subject of fertilizers and fertilizing materials in the manner followed in the first volume with soils. The general principles of fertilizer manufacture and application have been presented in so far as they seemed to throw light on the rational method of examination and analysis. The standard methods of analysis in use in this and other countries, have been presented with sufficient fullness for the guidance of the skilled worker, and the information of the student. To those who make use of a book only for routine work or for preparation for an examination, this volume, as its predecessor, will be found to have little attraction. This fact, however, will not be a cause of regret to the author whose purpose has been, avowedly, to present to the busy worker and student a broad view of a great subject which each one does not have the time to search out for himself.
It is a matter of regret, however, that the contents of the volume have again exceeded all expectations. It was found impracticable to secure any greater condensation without departing from the purpose, and impairing the completeness of the work. When work is done with no prospect of financial compensation, it is gratifying to find it appreciated, and the author will be content to have this volume meet with as kindly a reception as has been accorded volume one.
Washington, D. C.,
End of July, 1895.
[Pg iv]
TABLE OF CONTENTS OF
VOLUME SECOND.
PART FIRST.
PHOSPHATES AND PHOSPHATIC FERTILIZERS.
Introduction, pp. 1-4.—Natural fertilizers; Waste matters as fertilizing materials; Valuation of fertilizing ingredients.
General Analytical Processes, pp. 4-15.—Taking samples; Fertilizing minerals; Mixed fertilizers; French methods of taking samples; Sampling stable manures; Preparation of sample in laboratory; French and German methods; Drying fertilizer samples; Moisture in acid phosphates.
Analysis of Mineral Phosphates, pp. 15-39.—Constituents to be determined; Direct estimation of phosphoric acid; Official method; Preparation of solution; Use of tartaric acid; Determination of water and organic matters; Carbon dioxid; Soluble and insoluble matter; Silica and insoluble bodies; Estimation of lime; Ammonium oxalate method; Immendorff method; Estimation of iron and alumina; The acetate and Hess methods; Methods of Jones and Crispo; Geological Survey method; Method of Marioni and Fasselli; Method of Krug and McElroy; Method of Wyatt; Estimation of magnesia; of sulfuric acid; of fluorin.
General Methods for Phosphoric Acid, pp. 39-57.—Preliminary considerations; Estimation as stannic phosphate; Water soluble acid; Citrate insoluble acid; Total phosphoric acid; Norwegian methods; German experiment station methods; Soluble phosphoric acid; Swedish methods; Dutch methods; Errors in molybdate method; Color of pyrophosphate; Solution in sulfuric acid.
The Citrate Method, pp. 57-70.—General principles; Halle method; Swedish method; Methods adopted at the Brussels Congress; Dutch method for citrate soluble acid; Comparative accuracy of citrate and molybdate methods.
Basic Phosphatic Slags, pp. 70-86.—History and manufacture; Composition; Molecular structure; Solubility; Separation and solution; Estimation of total acid; Alternate method; Halle method of analysis; Dutch method; Estimation of citrate soluble acid in basic slags; Wagner’s shaking and digesting apparatus; Estimation of caustic lime in slags; Detection of adulteration.
Volumetric Determination of Phosphoric Acid, pp. 86-106.—Classification of methods; Uranium method; Preparation of sample; Precipitation of the phosphoric acid by magnesium citrate; [Pg v] Composition of magnesium citrate solution; Solution of ammonium magnesium phosphate; Preparation of standard solutions; Verifying standard solutions; Conduct of the analysis; Phosphoric acid in superphosphates; Determination of soluble and reverted phosphoric acid; Conclusions.
Titration of the Yellow Precipitate, pp. 106-118.—Pemberton’s method; Conduct of the analysis; Reactions; Calculation of results; Comparison with Official method; Titration as a lead compound; Water soluble acid; Estimation of phosphoric acid in presence of a large excess of iron; Emmerton method; Method of Dudley and Noyes; The Jones reductor; The volumetric silver method.
Technical Determination of Phosphoric Acid, pp. 118-125.—Desirability of methods; Reagents employed; Conduct of the molybdenum method; Conduct of the citrate method; Treatment of mineral phosphates and basic slags; Analysis of superphosphates.
Miscellaneous Notes on Phosphates and Phosphatic Fertilizers, pp. 126-150.—Time required for precipitation; Examination of the pyrophosphate; Iodin in phosphates; Chromium in phosphates; Estimation of Vanadium; Fluorin in bones; Note on separation of iron and alumina from phosphoric acid; Ammonium citrate soluble acid; Influence of time and strength of solvent on solution; Arbitrary Determination of reverted phosphoric acid; Digestion apparatus of Huston; Huston’s mechanical stirrer; Citrate method with small percentage of phosphoric acid; Direct precipitation of the citrate soluble phosphoric acid; Availability of phosphatic fertilizers; Direct weighing of the molybdenum precipitate.
Chemistry of the Manufacture of Superphosphates, pp. 150-156.—Reactions with phosphates; with fluorids; with carbonates; with iron and alumina; with magnesium compounds; Determination of quantity of sulfuric acid; Phosphoric acid superphosphates; Authorities cited in Part First.
PART SECOND.
NITROGEN IN FERTILIZERS AND
FERTILIZING MATERIALS.
Kinds of Nitrogen in Fertilizers, pp. 161-169.—Determination of state of combination; Microscopic examination; Seeds and seed residues; Fish scrap; Dried blood and tankage; Horn, hoof, and hair; Ammoniacal nitrogen; Nitrogen in guanos; Nitric nitrogen.
Methods of Analysis, pp. 169-192.—Classification of methods; Official methods; Combustion in copper oxid; Official volumetric method; Mercury pump; Combustion furnace; Process of combustion; Method of Johnson and Jenkins; Calculating results; Reading barometer; Tension of aqueous vapor; Aqueous tension in solutions of potassium hydroxid; Tables for calculating results; Soda-lime process; Official French [Pg vi] method; The ruffle soda-lime method; Official ruffle method; Boyer’s ruffle method.
Moist Combustion Process, pp. 192-220.—Historical; Method of Kjeldahl; Theory of the reactions; Preparation of reagents; Dutch kjeldahl method; Halle kjeldahl method; Official kjeldahl method; Distillation apparatus; Patrick’s distilling flask; Modification of the kjeldahl process; Method of Wilfarth; Method of Asboth; Method of Jodlbaur; Dutch jodlbaur method; Halle jodlbaur method; Official method for nitric nitrogen; Method of Scovell; Gunning method; Reactions of the gunning method; Official gunning method; Gunning method adapted to nitrates.
Determination of Nitrogen in Definite Forms of Combination, pp. 221-231.—Introductory considerations; Nitrogen as ammonia; Method of Boussingault; Determination of thiocyanates; Separation of albuminoid nitrogen; Separation of nitric nitrogen; Separation of ammoniacal nitrogen; Ulsch method for mixed fertilizers; Method of Schlöesing-Wagner; Schmitt’s modified method; Krüger’s method.
Sodium Nitrate, pp. 231-247.—Functions of sodium nitrate; Commercial forms of Chile saltpeter; Percentage of nitrogen in Chile saltpeter; Adulteration of Chile saltpeter; The Halle zinc-iron method for estimating nitrogen in Chile saltpeter; French method; Gantter’s volumetric method; Method of difference; Application of Chile saltpeter to the soil; Taking samples of soil to determine nitric nitrogen; The nitrifiable solution; Quantity of Chile saltpeter per acre; Consumption of Chile saltpeter; Authorities cited in Part Second.
PART THIRD.
Potash in Fertilizing Materials and Fertilizers, pp. 248-266.—Introduction; Forms of potash; Organic sources of potash; Tobacco stems and waste; Cottonseed hulls and meal; Wood ashes; Fertilizing value of ashes; Sugar beet molasses; Residue of wineries; Destruction of organic matter; Ignition with sulfuric acid; Potash in mineral deposits; Occurrence and history; Changes in potash salts in situ; Kainit; Carnallit; Polyhalit; Krugit; Sylvin; Sylvinit; Kieserit; Schönit; Potassium sulfate; Potassium magnesium carbonate; Potash in factory residues; Quantity of potash salts used.
Methods of Analysis, pp. 266-289.—Classification of methods; Platinic chlorid method; Official method; Alternate official method; Solution of organic compounds; Factors for calculation; Halle potash method; Dutch method; Swedish method; Method of the German Kali syndicate; Method for high grade potash salts; Barium oxalate method; de Roode method for kainit; Calcium chlorid method; Rapid control method; Determination from [Pg vii] metallic platinum; Errors in platinum method; Effect of concentration on accuracy; Differences in form of crystals of potassium platinochlorid; Recovery of the platinum waste.
Estimation of Potash as Perchlorate, pp. 289-301.—General principles; Caspari’s method of preparing perchloric acid; Kreider’s method; Keeping properties of perchloric acid; The analytical process for determining potassium as perchlorate; Removal of sulfuric acid; Applicability of the process; Accuracy of the process; Authorities cited in Part Third.PART FOURTH.
Miscellaneous Fertilizers, pp. 302-324.—Classification; Forms of lime; Application of lime; Action of lime; Analysis of lime; Gypsum, or land plaster; Analysis of gypsum; Common salt; Green vitriol; Stall manures; Hen manure; Guanos and cave deposits; Official French method for phosphoric acid in guanos; Leather waste; Wood ashes; Analysis of wood ashes; Method of analysis used in this laboratory; Official method for estimating alkalies in wood ashes; Statement of results; Authorities cited in Part Fourth; Index.
ILLUSTRATIONS TO
VOLUME SECOND.
| Page. | ||
| Figure 1. | Apparatus for crushing mineral fertilizers | 5 |
| “ 2. | Plate grinder for minerals | 6 |
| “ 3. | Shaking apparatus for superphosphates | 60 |
| “ 4. | Shaking machine for ammonium magnesium phosphate | 64 |
| “ 5. | Rössler ignition furnace | 65 |
| “ 6. | Wagner’s digestion apparatus for slags | 79 |
| “ 7. | Jones’ reduction tube | 115 |
| “ 8. | Huston’s digesting apparatus | 142 |
| “ 9. | Huston’s mechanical stirrer | 145 |
| “ 10. | Mercury pump and azotometer | 174 |
| “ 11. | Moist combustion apparatus of the Halle agricultural laboratory | 201 |
| “ 12. | Distillation apparatus of Halle agricultural laboratory | 203 |
| “ 13. | Distilling apparatus | 208 |
| “ 14. | Schlöesing-Wagner apparatus | 229 |
| “ 15. | Halle nitric acid apparatus | 234 |
| “ 16. | Gantter’s nitrogen apparatus | 237 |
| “ 17. | Geological relations of the potash deposits near Stassfurt | 258 |
[Pg 1]
VOLUME SECOND.
EXAMINATION OF FERTILIZING MATERIALS,
FERTILIZERS, AND MANURES.
1. Introduction.—In the first volume the principal plant foods occurring in soils have been named and the methods of estimating them described. As fertilizers are classed those materials which are added to soils to supply supposed deficiencies in plant foods, or to render more available the stores already present. There is little difference between the terms fertilizer and manure. In common language the former is applied to goods prepared for the farmer by the manufacturer or mixer, while the latter is applied to the stores accumulated about the stables or made elsewhere on the farm. Thus it is common to speak of a barnyard or stall manure and of a commercial fertilizer.
One of the objects of the analysis of soils, as described in the first volume of this work, is to determine the character of the fertilizer which should be added to a field in order to secure its maximum fertility.
One purpose of the present part is to determine the fitness of offered fertilizing material to supply the deficiencies which may be revealed by a proper study of the needs of the soil.
2. Natural Fertilizers.—In the succession of geologic epochs which has marked the natural history of the earth there have been brought together in deposits of greater or less magnitude the stores of plant food unused by growing crops or which may once have been part of vegetable and animal organisms. Some of these deposits have been mentioned in the first volume, paragraphs 11, 12, and 18. [Pg 2]
For a full description of the extent and origin of these deposits the reader is referred to works on economic geology. These deposits are the chief sources of the commercial fertilizers which are offered to the farmers of to-day and to which the agricultural analyst is called upon to devote much of his time and labor. The methods of determining the chemical composition and agricultural value of these deposits, as practiced by the leading chemists of this country and Europe, will be fully set forth in the following pages.
3. Waste Matters as Fertilizing Materials.—In addition to the natural products just mentioned the analyst will be called on also to deal with a great variety of waste materials which, in the last few years, have been saved from the débris of factories and abattoirs, and prepared for use on the farm. Among these waste matters may be mentioned, bones, horns, hoofs, hair, tankage, dried blood, fish scrap, oil cakes, ashes, sewage, and sewage precipitates, offal of all kinds, leather scraps, and organic débris in general.
It is important, before beginning an analysis, to know the origin of the substances to be determined. As has already been pointed out in volume first the process which would be accurate with a substance of a mineral origin might lead to error if applied to the same element in organic combination. This is particularly true of phosphorus and potash. A simple microscopic examination will usually enable the analyst to determine the nature of the sample. In this manner, in the case of a phosphate, it would at once be determined whether it was bone, mineral, or basic slag. The odor, color, and general consistence will also aid in the determination.
4. Valuation of Fertilizing Ingredients.—Perhaps there are no more numerous and perplexing questions propounded to the analyst than those which relate to the value of fertilizing materials. There is none harder to answer. As a rule these questions are asked by the farmer, and refer to the fertilizers put down on his fields. In such cases the cost of transportation is an important factor in the answer. The farther the farmer is removed from the place of fertilizer manufacture the greater, as a rule, will be the cost. Whether the transportation is [Pg 3] over land or by water also plays an important part in the final cost. The discovery of new stores of fertilizing materials has also much to do with the price. This fact is especially noticeable in this country, where the price of crude phosphates at the mines has fallen in a few years from nearly six dollars to three dollars and forty-three cents per ton[1]. This decrease has been largely due to discoveries of vast beds of phosphatic deposits in Florida, North Carolina, Tennessee, and Virginia. The state of trade, magnitude of crops, and the vigor of commerce also affect, in a marked degree, the cost of the raw materials of commercial fertilizers.
5. Trade Values of Fertilizing Ingredients in Raw Materials and Chemicals.—The values proposed by the Massachusetts Experiment Station are given below.[2]
| Cents per pound. |
|||
| Nitrogen | in | ammonia salts, | 19 |
| “ | “ | nitrates, | 14½ |
| Organic | nitrogen | in | dry and fine-ground fish, meat, blood, | |
| and in high-grade mixed fertilizers, | 18½ | |||
| “ | “ | “ | cottonseed meal, linseed meal, | |
| and castor pomace, | 15 | |||
| “ | “ | “ | fine-ground bone and tankage, | 16½ |
| “ | “ | “ | fine-ground medium bone and tankage, | 15 |
| “ | “ | “ | medium bone and tankage, | 12 |
| “ | “ | “ | coarse bone and tankage, | 7 |
| “ | “ | “ | hair, horn shavings, and coarse | |
| fish scraps, | 7 |
| Phosphoric | acid | soluble in water, | 6 |
| “ | “ | soluble in ammonium citrate, | 5½ |
| “ | “ | in fine bone and tankage, | 5½ |
| “ | “ | in fine medium bone and tankage, | 4½ |
| “ | “ | in medium bone and tankage, | 3 |
| “ | “ | in coarse bone and tankage, | 2 |
| “ | “ | in fine-ground fish, cottonseed meal, | |
| linseed meal, castor pomace, and wood-ashes, | 5 | ||
| “ | “ | insoluble (in ammonium citrate) in | |
| mixed fertilizers, | 2 [Pg 4] | ||
| Potash | as | high-grade sulfate, and in mixtures | |
| free from muriate, | 5 | ||
| “ | “ | muriate, | 4½ |
The manurial constituents contained in feed stuffs are valued as follows:
| Organic nitrogen, | 15 |
| Phosphoric acid, | 5 |
| Potash, | 5 |
The organic nitrogen in superphosphates, special manures, and mixed fertilizers of a high grade is usually valued at the highest figures laid down in the trade values of fertilizing ingredients in raw materials; namely, eighteen and one-half cents per pound, it being assumed that the organic nitrogen is derived from the best sources; viz., animal matter, as meat, blood, bones, or other equally good forms, and not from leather, shoddy, hair, or any low-priced, inferior form of vegetable matter, unless the contrary is evident. In such materials the insoluble phosphoric acid is valued at two cents a pound. These values change as the markets vary.
The scheme of valuation prepared by the Massachusetts station does not include phosphoric acid in basic slags. By many experimenters the value of the acid in this combination, tetracalcium phosphate, is fully equal to that in superphosphates soluble in water and ammonium citrate. It would perhaps be safe to assign that value to all the phosphoric acid in basic slags soluble in a five per cent citric acid solution.
Untreated fine-ground phosphates, especially of the soft variety, so abundant in many parts of Florida, have also a high manurial value when applied to soils of an acid nature or rich in humus. On other soils of a sandy nature, or rich in calcium carbonate, such a fertilizer would have little value. The analyst in giving an opinion respecting the commercial value of a fertilizer, must be guided not only by the source of the material, its fineness or state of decomposition, and its general physical qualities, but also by the nature of the crop which it is to nourish and the kind of soil to which it is to be applied.
[Pg 5] 6. Taking Samples.—It is impracticable to give definite directions for taking samples of fertilizers which will be applicable to all kinds of material and in all circumstances. If the chemist himself have charge of the taking of the sample, it will probably be sufficient to say that it should accurately represent the total mass of material sampled. Generally the samples which are brought to the chemist have been taken without his advice or direction and he is simply called upon to make an analysis of them.
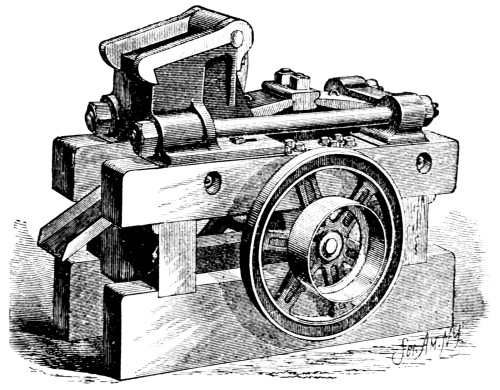
Figure. 1.
Apparatus for Crushing Mineral Fertilizers.
7. Minerals Containing Fertilizing Materials.—When possible, the samples should be accompanied by a description of the mines where they are procured and a statement of the geologic conditions in which the deposits were made. As large a quantity of the material as can be conveniently obtained and transported should be secured. Where a large quantity of mineral matter is at hand it should first be put through a crusher. Many forms of crusher, driven by hand and other power, are on the market. Among these may be mentioned the Alden, Blake, Bisworth, Forster, and Lipsay machines.[3] They are all constructed essentially on the same principle, the pieces of mineral being broken into small fragments between two heavy [Pg 6] vibrating steel plates. The general form of these instruments is seen in Fig. 1.
The fragments coming from the crusher can be reduced to a coarse powder by means of the iron plate and crusher shown in Fig. 2.
Where only a small quantity of mineral is at hand the apparatus just mentioned may be used at once after breaking the sample into small fragments by means of a hammer.
Finally the sample, if to be dissolved in an acid or soluble materials only, is reduced to a powder in an iron mortar until it will pass a sieve with a one or, better, one-half millimeter circular mesh. The powder thus obtained must be stirred with a magnet to remove all iron particles that may have been incorporated with the mass by abrasion of the instruments employed.
If a complete mineral analysis of the sample is to be secured, the material freed from iron, as above described, is to be rubbed to an impalpable powder in an agate mortar.
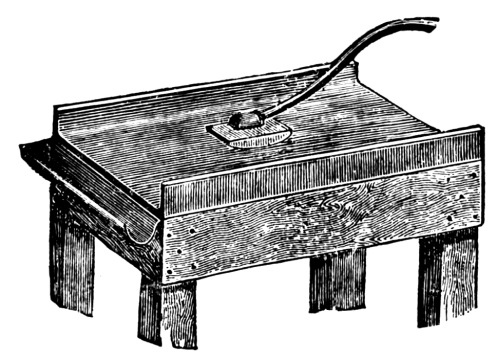
Figure. 2.
Plate Grinder for Minerals.
8. Mixed Fertilizers.—In fertilizing materials in bulk, the first requisite is that they shall be thoroughly mixed so that a given volume of the material may represent, practically, definite quantities of the materials sampled. The finer the material is, in the original state, and the more thoroughly it has been mixed, the better the sample will be. If the sample be already in sacks it will be sufficient to take portions by means of the ordinary trier, such as is used for [Pg 7] sampling sugar and other substances. This consists of a long metal implement such as would be formed by a longitudinal section of a tube. The end is pointed and suited for penetrating into the sack and the materials contained therein. On withdrawing it, the semi-circular concavity is found filled with the material sampled. Samples in this way should be taken from various parts of the sack and these samples well mixed together and a subsample of the amount necessary to be taken to the laboratory can then be obtained.
9. Method of the French Experiment Stations.—In the method employed by the French Experiment Stations it is directed that in no case should stones or other foreign particles be removed from the fertilizer sampled, but they should enter into the sample taken in, as nearly as possible, the same proportions as they exist in the whole mass.
In the case of stones or other solid masses which are to be sampled, as many samples as possible should be taken from all parts of the heap and these should be reduced to a coarse powder, thoroughly mixed together and sampled.
In case the material is in the form of a paste, if it is homogeneous, it will be sufficient to mix it well and take the sample directly; but in case there is a tendency for the pasty mass to separate into two parts, of which the one is a liquid and the other, more of a solid consistence, it may be well to take samples from each in case they can not be thoroughly incorporated by stirring.
10. Method of the French Association of Sugar Chemists.—The method adopted by the French sugar chemists directs that the sample should be taken from the fertilizer in bulk or from a portion used for industrial purposes.[4] The sample for analysis is to be taken from the above sample after it has been sent to the laboratory. The method of taking should be varied according to the condition of the substances to be analyzed.
The large sample selected from the goods delivered to commerce having been delivered at the laboratory, the analytical sample is taken as follows:
When the industrial sample, more or less voluminous, reaches the laboratory, the chemist is to begin by taking a note of the marks, labels, and descriptions found thereon, and of the nature and state of the package which contains it, and the date of its arrival. All this [Pg 8] information should be entered upon the laboratory book and afterwards transcribed on the paper containing the results of the analysis, as well as the name of the person sending it. This having been done, the sample is to be properly prepared in order that a portion may be taken representing exactly the mean composition of the whole.
If it is in a state of fine powder, such as ground phosphates and certain other fertilizers, it is sufficient to pass it two or three times through a sieve with meshes one millimeter in diameter, taking care to break up the material each time in order to mix it and to pulverize the fragments which the sieve retains. The whole is afterwards spread in a thin layer upon a large sheet of paper, and a portion is taken here and there upon the point of a knife until about twenty grains are removed, and from this the portion subjected to analysis is afterwards taken.
If the sample comes in fragments, more or less voluminous, such as phosphatic rocks or coarsely pulverized guanos containing agglomerated particles, it is necessary first to reduce the whole to powder by rubbing it in a mortar or in a small drug mill. It is next passed through a sieve of the size mentioned above and that which remains upon the sieve pulverized anew until all has passed through. This precaution is very important, since the parts which resist the action of the pestle the most have often a composition different from those which are easily broken.
When the products to be analyzed contain organic materials, such as horn, flesh, dry blood, etc., the pulverization is often a long and difficult process, and results in a certain degree of heating which drives off some of the moisture in such a way that the pulverized product is at the last drier, and, consequently, richer than the primitive sample. It is important to take account of this desiccation, and since the pulverization of a mass so voluminous can not be made without loss, the determination of the total weight of the sample before and after pulverization does not give exact results.
In such a case it is indispensable to determine the moisture, both before and after pulverizing, and to calculate the analytical results obtained upon the pulverized sample back to the original sample.
In order to escape this necessity, as well as the difficulties resulting from the variations in moisture during transportation, some [Pg 9] chemists have thought it better to always dry the commercial products before submitting them to analysis, and to report their results in the dry state, accompanied by a determination of the moisture, leaving thus to the one interested the labor of calculating the richness in the normal state, that is to say, in the real state in which the merchandise was delivered.
In addition to the fact that this method allows numerous chances of errors, many substances undergoing important changes in their composition by drying alone, it has been productive of the most serious consequences. The sellers have placed their wares on the market with the analysis of the material in a dry state, and a great number of purchasers have not perceived the fraud concealed under this expression so innocent in appearance. It is thus that there has been met with in the markets guano containing twenty-five per cent of water, which was guaranteed to contain twelve per cent of phosphoric acid, when, in reality, it contained only eight per cent in the moist state.
11. Barn-Yard Manures.—The sampling of stall and barnyard manures is more difficult on account of the fact that the materials are not homogeneous and that they are usually mixed with straw and other débris from the feed trough, and only the greatest care and patience will enable the operator to secure a fair sample.
In the case of liquid manures the liquid should be thoroughly stirred before the sample is taken.
Frear points out the difficulty of securing representative samples of stall manure and describes methods of removing it.[5] The stall manure sampled had been piled in the cattle-yard for a time and the cattle were allowed to run over the heaps for an hour or two each day. Pigs were also allowed free access to the heaps in order to insure a more perfect mixture of the ingredients.
Twenty-nine loads of 3,000 pounds each were taken from the exposed heap and thirty-four loads of 2,000 pounds each were taken from the covered heap. From each load were removed two carefully selected portions of ten pounds each, which were placed in separate covered boxes numbered A and B. When the sampling was completed these boxes were covered. After being removed to the laboratory the boxes were weighed and the contents [Pg 10] thoroughly mixed. Two samples of twelve liters volume each, were drawn from each box. One-third of this was chopped in a large meat chopper and the other two-thirds taken into the laboratory without being cut. These samples, on entering the laboratory, were weighed and dried at a temperature of 60°. Smaller samples were then drawn from each of these and ground in a drug mill for analysis. Duplicate samples taken in this way, while they did not give absolutely concordant results, showed a near approximation. A more careful sampling on the line proposed would, in all probability, secure absolutely agreeing results in duplicate samples.
12. Preparation of Sample in Laboratory.—The method of preparing mineral fertilizers for analysis has been given under directions for sampling. Many difficulties attend the proper preparation of other samples, and the best approved methods of procedure are given below:
According to the directions given by the Association of Official Agricultural Chemists the sample should be well intermixed, finely ground, and passed through a sieve having circular perforations one millimeter in diameter.[6] The processes of grinding and sifting should take place as rapidly as possible so that there may be no gain or loss of moisture during the operation.
13. Method of the French Agricultural Stations.—The manner of proceeding recommended by the French stations varies with the fertilizer.[7] If it is not already in the form of a powder it is necessary to pulverize it as finely as possible by rubbing it up in a mortar. In certain cases, as with superphosphates, the material should be passed through a sieve having apertures of one millimeter diameter, all the larger parts being pulverized until they will pass this sieve.
When the matters are too pasty to be divided in the mortar they should be divided by means of a knife or a spatula. They should then be incorporated with a known weight of inert, pulverulent matter such as fine sand, with which they should be thoroughly mixed and in subsequent calculations the quantity of sand or other inert matter added must be taken into consideration. Usually a pasty state of a fertilizer is due to the humidity of the mixture. In this case a considerable volume of [Pg 11] the sample is taken and dried and then reduced to a pulverulent state. In the subsequent calculations, however, the percentage of moisture lost must be taken into consideration.
Before drying a sample it is necessary to take into consideration whether or not the product will be modified by desiccation as would be the case, for instance, with superphosphates. With these, which are often in a state more or less agglomerated, it is recommended to introduce into them, in order to divide them, a certain quantity of calcium sulfate in order to obtain them in a pulverulent state.
In the case of animal débris they should be divided as finely as possible with the aid of scissors and then passed through a drug mill if dry enough. They are then mixed by hand and may finally be obtained in a state of considerable homogeneity.
When fertilizers are in a pasty state more or less liquid, they are dried at 100°, first introducing a little oxalic acid in case they contain any volatile ammoniacal compounds. The product of desiccation is then passed through a mill. Before treating in this way it is necessary to be sure that the composition will not be altered by drying. In the case of a mixture containing superphosphates and nitrate, for instance, drying would eliminate the nitric acid. In such a case the free phosphoric acid should be neutralized with a base like lime. In the case of fertilizers containing both nitrates and volatile ammoniacal compounds the addition of oxalic acid might also set free nitric acid during the desiccation. In such a case it is necessary to dry two samples; one with the addition of oxalic acid for the purpose of estimating the ammonia, and the other without the acid for the purpose of estimating the nitrate. A qualitative analysis should precede all the operations so as to determine the nature of the material to be operated on.
14. German Method.—In the method pursued by the German experiment stations it is directed:[8]
(1) Dry samples of fertilizers must be passed through a sieve and afterwards well mixed.
(2) With moist fertilizers, which can not be subjected to the above process, the preparation should consist in a careful and thorough mixing, without sieving. [Pg 12]
(3) On the arrival of the samples in the laboratory their weight should be determined. The half of the sample is prepared for analysis and the other part, to the amount of a kilo, should be placed in a glass vessel, closed air-tight, and placed in a cool place for at least a quarter of a year from the time of its reception, in order that it may be subjected to any subsequent investigations which may be demanded.
(4) In the case of raw phosphates and bone-black the amount of water which they contain should be determined at from 105° to 110°. Samples which in drying lose ammonia in any way, should have this ammonia determined.
(5) Samples which are sent to other laboratories for control analyses, should be sent securely packed in air-tight glass bottles.
(6) The weight of the samples sent should be entered in the certificates of analysis.
(7) Samples which, on pulverizing, change their content of water, must have the water content estimated in both the coarse and powdered condition and the results of the analysis must be calculated to the water content of the original coarse substance.
15. Special Cases.—Many cases arise of such a nature as to make it impossible to lay down any rule which can be followed with success. As in almost every other process in agricultural chemistry the analyst in such cases must be guided by his judgment and experience. Keeping in view the main object, viz., to secure in a few grams of material a fair representation of large masses he will generally be able to reach the required result by following the broad principles already outlined. In many cases the details of the work and the adaptations necessary to success must be left to his own determination.
16. Drying Samples of Fertilizers.—The determination of the uncombined moisture in a sample of fertilizer is not an easy task. In some cases, as in powdered minerals, drying to constant weight at the temperature of boiling water is sufficient. In organic matters containing volatile nitrogenous compounds, these must first be fixed by oxalic or sulfuric acid, before the desiccation begins. If any excess of sulfuric acid be added, however, drying at 100° becomes almost impossible. Particular precautions must be observed in drying [Pg 13] superphosphates. In drying samples preparatory to grinding for analysis, it is best to stop the process as soon as the materials can be pulverized. In general, samples should be dried only to determine water, and the analytical processes should be performed on the undried portions. It is not necessary, as a rule, to dry samples of fertilizers in an inert atmosphere, such as hydrogen or carbon dioxid. Drying in vacuo may be practiced when it is desired to secure a speedy desiccation or one at a low temperature.
17. Official Methods.—The Official Agricultural Chemists direct, in the case of potash salts, sodium nitrate, and ammonium sulfate, to heat from one to five grams in a flat platinum or aluminum dish at 130° until the weight is constant.[9] The loss in weight is taken to represent the water. In all other cases heat two grams, or five grams if the sample be very coarse, for five hours in a steam-bath.
In the German stations in the case of untreated phosphates and bone-black the moisture is estimated at from 105° to 110°. Samples which lose ammonia should have the weight of ammonia given off at that temperature, determined separately.
For purposes of comparison it would be far better to have all contents of moisture determined at the boiling-point of water. While this varies with the altitude and barometric pressure yet it is quite certain that the loss on drying to constant weight at all altitudes is practically the same. Where the atmospheric pressure is diminished for any cause the water escapes all the more easily. This, practically, is a complete compensation for the diminished temperature at which water boils.
Where the samples contain no ingredient capable of attacking aluminum, they can be conveniently dried, in circular dishes of this metal about seven centimeters in diameter and one centimeter deep, to constant weight, at the temperature of boiling water.
18. Moisture in Monocalcium Phosphates.—In certain fertilizers, especially superphosphates, containing the monocalcium salt, the estimation of water is a matter of extreme difficulty on account of the presence of free acids and of progressive changes in the sample due to different degrees of heat. [Pg 14]
Stoklasa has studied these changes and reaches the following results[10]:
A chemically pure monocalcium phosphate of the following composition, viz.,
| CaO | 22.36 | per | cent. |
| P₂O₅ | 56.67 | “ | “ |
| H₂O | 21.53 | “ | “ |
was subjected to progressive dryings. The loss of water after ten hours was 1.83 per cent; after twenty hours, 2.46 per cent; after thirty hours, 5.21 per cent; after forty hours, 6.32 per cent; after fifty hours, 6.43 per cent. This loss of water remained constant at 6.43 per cent. This loss represents one molecule of water as compared with the total molecular magnitude of the mass treated. A calcium phosphate, therefore, of the following composition, CaH₄(PO₄)₂·H₂O loses, after forty hours, drying at 100°, its water of crystallization. The calcium phosphate produced by this method forms opaque crystals which are not hygroscopic and which give, on analysis, the following numbers:
| CaO | 24.02 | per | cent. |
| P₂O₅ | 16.74 | “ | “ |
| H₂O | 15.09 | “ | “ |
The temperature can be raised to 105° without marked change. If the temperature be raised to 200° the decomposition of the molecule is hastened according to the following formula:
4 CaH₄(PO₄)₂ = Ca₂P₂O₇ + Ca(PO₃)₂ + CaH₂P₂O₇ + 2 H₃PO₄ + 4 H₂O.
The chemical changes during the drying of monocalcium phosphates can be represented as follows, temperature 200° for one hour:
8[CaH₄(PO₄)₂·H₂O] = 4CaH₄(PO₄)₂ + Ca(PO₃)₂ + Ca₂P₂O₇
+ CaH₂P₂O₇ + 2H₃PO₄ + 12H₂O.
The further drying at 200° produces the following decomposition:
4CaH₄(PO₄)₂ + Ca(PO₃)₂ + Ca₂P₂O₇ + CaH₂P₂O₇ + 2H₃PO₄
= 2Ca(PO₃)₂ + 4CaH₂P₂O₇ + Ca₂P₂O₇ + 2H₃PO₄ + 5H₂O.
2Ca(PO₃)₂ + 4CaH₂P₂O₇ + Ca₂PO₇ + 2H₃PO₄
= 6Ca(PO₃)₂ + 2CaH₂P₂O₇ + 5H₂O.
[Pg 15] Finally, pyrophosphate at 210° is completely decomposed into metaphosphate and water according to the following formula:
6Ca(PO₃)₂ + 2CaH₂P₂O₇ = 8Ca(PO₃)₂ + 2H₂O.
Provided the drying is made at once at 210° the sum of the changes produced as indicated above, can be represented by the following formula:
8[CaH₄(PO₄)₂·H₂O] = 8Ca(PO₃)₂ + 24H₂O.
19. Constituents to be Determined.—The most important point in the analysis of mineral phosphates is to determine their content of phosphoric acid. Of equal scientific interest, however, and often of great commercial importance is the determination of the percentage of other acids and bases present. The analyst is often called on, in the examination of these bodies, to make known the content of water both free and combined, of organic and volatile matter, of carbon dioxid, sulfur, chlorin, fluorin, silica, iron, alumina, calcium, manganese, magnesia, and the alkalies. The estimation of some of these bodies presents problems of considerable difficulty, and it would be vain to suppose that the best possible methods are now known. Especially is this the case with the processes which relate to the estimation of the fluorin, silica, iron, alumina, and lime. The phosphoric acid, however, which is the chief constituent from a commercial point of view, it is believed, can now be determined with a high degree of precision. Often the estimation of some of the less important constituents is of great interest in determining the origin of the deposits, especially in the case of fluorin. While the merchant is content with knowing the percentage of phosphoric acid and the manufacturer asks in addition only some knowledge of the quantity of iron, alumina, and lime the analyst in most cases is only content with a complete knowledge of the constitution of the sample at his disposal.
20. Direct Estimation of the Phosphoric Acid.—It often happens, in the case of a mineral phosphate, that the only determination desired is of the phosphoric acid. In this instance the analyst may proceed as follows: If the qualitative test shows the usual amount of phosphoric [Pg 16] acid, two grams of the sample passed through a sieve, with a millimeter mesh, are placed in a beaker and thoroughly moistened with water. The addition of water is to secure an even action of the hydrochloric acid on the carbonates present. The beaker is covered with a watch-glass and a little hydrochloric acid is added from time to time until all effervescence has ceased. There are then added about thirty cubic centimeters of aqua regia and the mixture raised to the boiling-point on a sand-bath or over a lamp. The heating is continued until chlorin is no longer given off and solution is complete. The volume of the solution is then made up to 200 cubic centimeters without filtering, filtered, and an aliquot part of the filtrate, usually fifty cubic centimeters, representing half a gram of the original sample, taken for the determination of the phosphoric acid according to the method of the Official Agricultural Chemists. The small quantity of insoluble material does not introduce any appreciable error into the process when the volume is made up to 200 or 250 cubic centimeters.
21. Method of the Official Agricultural Chemists for Total Phosphoric Acid.—To the hot solution, for every decigram of phosphorus pentoxid which may be present, add fifty cubic centimeters of the molybdic solution. Digest at 65° for an hour, filter, and wash with water or ammonium nitrate solution[11]. Test the filtrate by renewed digestion with additional molybdate reagent. Dissolve the precipitate on the filter with ammonia in hot water and wash into a beaker, making the volume of filtrate and washings not more than 100 cubic centimeters. Nearly neutralize with hydrochloric acid, cool, and add magnesia mixture from a burette at the rate of about one drop a second, stirring vigorously, meanwhile. The quantity of magnesia mixture to be added is not prescribed in the official method but it should always be in excess of the amount necessary for complete precipitation. For each decigram of phosphorus pentoxid, from eight to ten cubic centimeters should be used. Fifteen minutes after the last of the magnesia mixture has been stirred in, thirty cubic centimeters of ammonia of 0.95 specific gravity are added and the beaker set aside for two hours or longer. The ammonium magnesium phosphate is separated by [Pg 17] filtration, dried, ignited gently at first, and finally over a blast-lamp and weighed as magnesium pyrophosphate. The factors for calculating the phosphorus pentoxid and tricalcium phosphate from the weight of pyrophosphate are given below on the two bases; viz., hydrogen equals 1, and oxygen equals 16.
H = 1.
Mg₂P₂O₇ × 0.63976 = P₂O₅
Mg₂P₂O₇ × 1.3964 = Ca₃(PO₄)₂
P₂O₅ × 2.1827 = Ca₃(PO₄)₂
O = 16.
Mg₂P₂O₇ × 0.63792 = P₂O₅
Mg₂P₂O₇ × 1.3926 = Ca₃(PO₄)₂
P₂O₅ × 2.1831 = Ca₃(PO₄)₂
22. Preparation of Solutions.—Molybdic Solution.—Dissolve 100 grams of molybdic acid in 400 grams or 417 cubic centimeters of ammonia, of 0.96 specific gravity, and pour the solution thus obtained into 1,500 grams or 1,250 cubic centimeters of nitric acid, of 1.20 specific gravity. Keep the mixture in a warm place for several days, or until a portion heated to 40° deposits no yellow precipitate of ammonium phosphomolybdate. Decant the solution from any sediment and preserve in glass-stoppered vessels.
Magnesia Mixture.—Dissolve twenty-two grams of recently ignited calcined magnesia in dilute hydrochloric acid, avoiding an excess of the latter. Add a little calcined magnesia in excess, and boil a few minutes to precipitate iron, alumina, and phosphoric acid; filter, add 280 grams of ammonium chlorid, 700 cubic centimeters of ammonia of specific gravity 0.96, and water enough to make a volume of two liters. Instead of the solution of twenty-two grams of calcined magnesia, 110 grams of crystallized magnesium chlorid may be used.
Dilute Ammonia for Washing.—One volume of ammonia, of 0.96 specific gravity, mixed with three volumes of water, or usually one volume of concentrated ammonia with six volumes of water.
23. Use of Tartaric Acid in Phosphoric Acid Estimation.—In the presence of iron the molybdate mixture is likely to carry down some ferric oxid with the yellow precipitate. To prevent this, and also hinder the separation of molybdic acid in the solution on long standing, tartaric acid has been recommended.
Jüptner has found that the presence of tartaric acid does not interfere [Pg 18] with the separation of the yellow precipitate, as some authorities assert.[12] Even 100 grams of the acid in one liter of molybdate solution produce no disturbing effect. Molybdate solution treated with tartaric acid did not show any separation of molybdic acid when kept for a year at room temperatures. The presence of tartaric acid, therefore, is highly recommended by him to prevent the danger of obtaining both ferric oxid and molybdic acid with the yellow precipitate.
24. Water and Organic Matters.—The sample, according to the practice of Chatard, should be ground fine enough to leave no residue on an eighty mesh sieve, and should be thoroughly mixed by passing it three times through a forty mesh sieve[13].
Two grams are weighed into a tared platinum crucible. This, with its lid, is placed in an air-bath at 105°, and heated for at least three hours. The lid is then put on, and the crucible is placed in a desiccator and weighed as soon as cold. The loss in weight is the moisture.
Wyatt recommends that two grams of the fine material be heated in ground watch-glasses, the edges of which are separated so as to allow the escape of the moisture.[14] The heating is continued for three hours at 110°, the watch-glasses then closed and held by the clip, cooled in a desiccator, and weighed. This method is excellent for very hygroscopic bodies, but where quick-acting balances are used, scarcely necessary for a powdered mineral.
The residue from the moisture determination is gradually heated to full redness over a bunsen, and then ignited over the blast-lamp. This operation is repeated after weighing until a constant weight is obtained. The loss (after deducting the percentage of carbon dioxid as found in another portion) may be taken as water and organic matter. This method is sufficient for all practical purposes; but when minerals containing fluorin are strongly ignited, a part of the fluorin is expelled; hence, if more accurate determinations are required, the loss of fluorin must be taken into account. In this laboratory it has been proved that a pure calcium fluorid undergoes progressive decomposition at a bright red heat with formation of lime.
Wyatt directs that the combined water and organic matters be determined in the residue from the moisture estimation as follows: The residue is brushed into a weighed platinum crucible, which is heated over a small bunsen for ten minutes and then brought to full heat of a blast-lamp for five minutes. After cooling, the total loss is determined by weighing. After deducting the carbon dioxid determined in a separate portion, the residual loss is regarded as due to combined moisture and organic matter.
25. Carbon Dioxid.—Many forms of compact apparatus have been devised for this estimation, but none of them is satisfactory if accurate results are desired.[15] Not to mention other objections, many phosphates must be heated nearly to the boiling-point with dilute acid to effect complete decomposition of the carbonates. The distillation method described by Gooch[16] is excellent, and when once the apparatus is set up, its work will be found to be rapid and satisfactory.
Wyatt regards the estimation of carbon dioxid as one of the most [Pg 19] important for factory use. The carbonates present in a sample indicate the loss of an equivalent amount of acid in the process of conversion into superphosphate.[17]
The apparatus employed for estimating carbon dioxid may be any one of those in ordinary use for this purpose. The principle of the process depends on the liberation of the gas with a mineral acid, its proper desiccation, and subsequent absorption by a caustic alkali, best in solution.
The apparatus of Knorr, described in volume first, page 338, may be conveniently used. The weight of the sample to be used should be regulated by the content of carbonate. When this is very high, from one to two grams will be found sufficient; when low, a larger quantity must be used. Hydrochloric is preferred as the solvent acid. Those forms of apparatus which are weighed as a whole and the carbon dioxid determined by reweighing after its expulsion, are not as reliable as the absorption apparatus mentioned.
26. Soluble and Insoluble Matter.—Five grams of the fine phosphate are put into a beaker, twenty-five cubic centimeters of nitric acid, (specific gravity 1.20) and 12.5 cubic centimeters of hydrochloric acid (specific gravity 1.12) are added. The beaker, [Pg 20] covered with a watch-glass, is placed upon the water-bath for thirty minutes[18]. The contents of the beaker are well stirred from time to time, and at the end of the period the beaker is removed from the bath, filled with cold water, well stirred, and allowed to settle. The solution is next filtered into a half liter flask, and the residue is thoroughly washed with cold water, partially dried, and then ignited, (finishing with the blast-lamp) and brought to constant weight. The figures thus obtained will, however, be incorrect, because the fluorin liberated during the solution of the phosphates dissolves a portion of the silica. Hence, the results are too low. Nevertheless, as the same action would occur in the manufacture of a superphosphate from the material, the determination may be considered, as a fair approximation to commercial practice. The ignited residue must be tested for phosphorus pentoxid.
27. Preparation of the Solution.—The flask containing the filtrate is filled to the mark with cold water, and the solution is thoroughly mixed by twice pouring into a dry beaker and returning it to the flask. Cold water is used for washing the residue, since if hot water be used, the sesquichlorids are apt to become basic and insoluble, and hence to remain in the residue and on the filter paper. Besides, as the flask is to be filled to the mark, the contents must be cold before any volumetric measurements can be made.
28. Silica and Insoluble Bodies.—Wyatt describes the following method for determining the total insoluble or siliceous matters in a mineral phosphate[19]. Five grams of the fine sample are placed in a porcelain dish with about thirty cubic centimeters of aqua regia. The dish is covered with a funnel, placed on a sand-bath and, after solution is complete, evaporated to dryness with care to prevent sputtering. When dry the residue is moistened with hydrochloric acid and again dried, rubbing meanwhile to a fine powder. The heat of the bath is then increased to 125° and maintained at this temperature for about ten minutes. When cool, the residue is treated with fifty cubic centimeters of hydrochloric acid for fifteen minutes. The acid is then diluted and filtered on a gooch, which is washed with hot water until the filtrate amounts to a quarter of a liter. The residue in the crucible is dried, [Pg 21] ignited, and weighed. This method, unless the solution be subsequently boiled with nitric acid, may not retain all the phosphoric acid in the ortho form.
It is difficult to estimate the total silica by the ordinary methods of mineral analysis. This is due to the fact that in an acid solution of a substance containing silicates and fluorids the whole of the silica or the fluorin, as the case may be may escape as silicofluorid on evaporation. Again, it is not easy to decompose calcium phosphate by fusing with sodium carbonate. If an attempt be made to do this, however, the process should be conducted as follows: A portion of the sample is ground to an impalpable powder in an agate mortar. From one to two grams of the substance are mixed with five times its weight of sodium carbonate and fused with the precautions given in standard works on quantitative analysis. The fused mass is digested in water, boiled, and filtered, and the residue washed first with boiling water and afterwards with ammonium carbonate. The filtrate contains all the fluorin as sodium fluorid and, in addition to this, sodium carbonate, silicate, and aluminate. Mix the filtrate with ammonium carbonate and heat for some time, replacing the ammonium carbonate which evaporates. Separate by filtration the silicic acid hydrate and aluminum hydroxid which are formed and wash them with ammonium carbonate. To separate the last portions of silica from the filtrate, add a solution of zinc oxid in ammonia. Evaporate until no more ammonia escapes and separate, by filtration, the zinc silicate and oxid. Determine the silica in this precipitate by dissolving in nitric acid, evaporating to dryness, taking up with nitric acid and separating the undissolved silica by filtration. In the alkaline filtrate the fluorin may be estimated by the usual method as calcium salt.
29. Estimation of Lime.—One hundred cubic centimeters of the solution (containing one gram of the original substance) are evaporated in a beaker to about fifty cubic centimeters; ten cubic centimeters of dilute sulfuric acid (one to five) are added; and the evaporation is continued on the water-bath until a considerable crop of crystals of gypsum has formed[20]. The solution is then allowed to cool, when it generally becomes pasty, owing to the separation of additional gypsum. When it is cold, 150 [Pg 22] cubic centimeters of ninety-five per cent alcohol are slowly added, with continual stirring, and the whole is allowed to stand for three hours, being stirred from time to time. After three hours, it is filtered, with the aid of a filter-pump, into a distillation flask, and the beautifully crystalline precipitate, which does not adhere to the beaker, is washed with ninety-five per cent alcohol. The filter, with the precipitate, is gently removed from the funnel and inverted into a platinum crucible, so that, by squeezing the point of the filter, the precipitate is made to fall into the crucible, and the paper can be pressed down smoothly upon it. On gentle heating of the crucible, the remaining alcohol burns off, and when the paper has been completely destroyed, the heat is raised to the full power of a bunsen for about five minutes. After cooling in a desiccator the crucible containing the calcium sulfate, is weighed. The filtration may also be accomplished on asbestos felt.
30. The Ammonium Oxalate Method.—This method has been extensively used in this country in commercial work, and is best carried out as described by Wyatt.[21] The total filtrates from the iron and alumina precipitates, secured as described in paragraph 33, are well mixed and concentrated to a volume of about 100 cubic centimeters. There are added about twenty cubic centimeters of a saturated solution of ammonium oxalate, and after stirring, the mixture is allowed to cool and remain at rest for six hours. The supernatant liquid is poured through a filter, the residue washed three times by decantation with hot water and brought upon the filter. The beaker and precipitate are washed at least three times. The precipitate is dried and ignited at low redness for ten minutes. The temperature is then raised by a blast and the ignition continued for five minutes longer, or until the lime is obtained as oxid. The precipitate is likely to contain magnesia. The magnesia is estimated in the filtrates from the lime determination by first mixing them and concentrating to 100 cubic centimeters, which, after cooling, are made strongly alkaline with ammonia. After allowing to stand for twelve hours the ammonium magnesium phosphate is collected and reduced to magnesium pyrophosphate by the usual processes. If one gram of the original material has been used the pyrophosphate obtained, multiplied by 0.36, will give the weight of magnesia contained therein. [Pg 23]
31. Lime Method of Immendorff.—The tedious processes required to determine the lime in the presence of iron, alumina, and large quantities of phosphoric acid are well known to analysts. Immendorff has published a method, accompanied by the necessary experimental data, based on the comparative insolubility of calcium oxalate in very dilute solution of hydrochloric acid. He has shown in the data given that the lime is all precipitated in the conditions named and that the precipitate, when properly prepared, is not contaminated with weighable amounts of the other substances found in the original solution[22]. The ease with which oxalic acid can be determined volumetrically with potassium permanganate solution aids greatly in the time-saving advantages of the process.
In a hydrochloric acid solution of a mineral phosphate an aliquot part of the filtrate representing about 250 milligrams of calcium oxid, usually about twenty-five cubic centimeters, should be taken for the analysis. Ammonia is added in slight excess and then the acid reaction restored with hydrochloric until shown plainly by litmus. The solution is then heated and the lime thrown down by adding a solution of ammonium oxalate in excess. In order to secure a greater dilution of the hydrochloric acid after the precipitation has been made, water should be added until the volume is half a liter. Before filtering, the whole should be cooled to room temperature. The precipitate should be washed first with cold and afterwards with warm water. The well-washed precipitate is dissolved in hot dilute sulfuric acid and the solution, while hot, titrated with a standard solution of potassium permanganate set by a solution of ammonio-ferrous sulfate.
If one cubic centimeter of the permanganate represent 0.005 gram of iron it will correspond almost exactly to 0.0035 gram of calcium oxid.
Example.—Sample of rather poor mineral phosphate, five grams in half a liter. Strength of potassium permanganate, one cubic centimeter equivalent to 0.00697 gram of iron and to 0.003484 gram of calcium oxid.
Twenty-five cubic centimeters of the solution, representing one quarter of a gram, in which the lime was precipitated as above described, required 9.6 cubic centimeters of the potassium permanganate to saturate the oxalic acid. Then
9.6 × 0.003484 = 0.0334464 gram,
or 13.38 per cent of calcium oxid. The method is also applicable to basic slags.
[Pg 24] 32. Estimation of Iron and Alumina in Mineral Phosphates.—When mineral phosphates are to be used for the manufacture of superphosphates by treatment with sulfuric acid their content of iron and alumina becomes a matter of importance. By reason of the poor drying qualities of the sulfates of these bases their presence in any considerable excess of a few per cent becomes exceedingly objectionable. The accurate estimation of these ingredients is not only then a matter of scientific interest but one of great commercial significance to the manufacturer.
The conventional methods so long in use depending on the precipitation of the iron and alumina as phosphates in the presence of acetic acid have been proved to be somewhat unreliable. Not only does the acetic acid fail to prevent the precipitation of some of the lime, but it also dissolves more or less of the iron and aluminum phosphates. The solution of the precipitate and its reprecipitation by the addition of ammonia, may free the second precipitate from lime, but it increases the error due to the solubility of the aluminum salt. The methods recently introduced for the estimation of iron and alumina in presence of excess of lime and phosphoric acid are not entirely satisfactory, but are the best which can now be offered.
33. The Acetate Method.—The principle of this process is based on the fact that in a solution containing iron, alumina, lime, and phosphoric acid the iron and aluminum phosphates can be thrown down in a slightly acid solution by ammonium acetate while the calcium phosphate remains in solution. The acidity in the older methods is due to acetic and can be secured by making the solution slightly alkaline with ammonia and adding acetic to slight acidity. One of the best methods of conducting the operation is that of C. Glaser[23]. Glaser’s modification of the older processes is based on the assumption that at 70° the aluminum phosphate is quantitatively precipitated by ammonium acetate in a dilute hydrochloric acid solution and that the mixed precipitates of iron and aluminum phosphates obtained at this [Pg 25] temperature are free of lime. The operation is conducted in the following manner:
The hydrochloric acid solution of the phosphate must contain no free chlorin and is treated with a few drops of a methyl orange solution. Ammonia is added until nearly neutral, but the acid reaction is retained as shown by the indicator. A few cubic centimeters of ammonium acetate are added, which produce a yellow coloration of the liquid and also a complete precipitation of the iron and aluminum phosphates when warmed to 70°. At this temperature the precipitation of any calcium phosphate is avoided. A small quantity of the lime may be carried down mechanically and therefore the precipitate should be dissolved in hydrochloric acid and the precipitation again made as above after the addition of some sodium phosphate. If the original solution contain any free chlorin, as may be the case when aqua regia is employed as solvent, before beginning the separation, ammonia should be added in slight excess and the acidity restored by hydrochloric after adding the indicator. In washing the precipitates, water of not over 70° must be used. As has been shown by Hess in the work cited in the next paragraph, the statement of C. Glaser to the effect that the precipitates obtained as above are free of lime has not been proved to be strictly correct. The process, however, is a distinct improvement over the older methods and forms the basis of the amended process given below, which appears to be sufficiently accurate to entitle the acetate method to favorable consideration.
34. Method of Hess.—Hess has lately made a thorough investigation of the standard methods of determining the iron and aluminum oxids in the presence of phosphoric acid and has shown that the assumption that the composition of the precipitate is represented by the formula Al₂(PO₄)₂ + Fe₂(PO₄)₂ is erroneous[24].
In the washing of the precipitated iron and aluminum phosphates there is a progressive decomposition of the compound with the production of a basic salt. The composition of the precipitate at the end is dependent chiefly upon the way in which the washing takes place. It is quite difficult to always secure a washing in exactly the same way and the [Pg 26] final composition of the precipitate varies with almost every determination. It is not, therefore, an accurate proceeding to take half the weight of the precipitate as phosphoric acid or as iron oxid and alumina. In every case it is necessary to dissolve the precipitate and determine the phosphoric acid in the regular way. Hess proposes the following method for carrying out the acetate process of separation:
The mineral phosphate should be dissolved in hydrochloric acid and the solution made up to such a volume as shall contain in each fifty cubic centimeters, one gram of the original substance. This quantity of the solution is diluted with two or three times its volume of water to which a drop of methyl orange solution (1-100) is added, and ammonia added with constant stirring until the solution is just colored and still reacts slightly acid. Without taking any account of the precipitate which is produced by this approximate neutralization of the solution, there are added fifty cubic centimeters of acid ammonium acetate which in one liter contains 250 grams of commercial ammonium acetate. The acidity of the solution is due to an excess of acetic in the commercial salt. The temperature is then carried to 70° and the precipitate produced immediately separated by filtration, washed four times with water below 70°, and again dissolved in dilute hydrochloric acid. The dissolved precipitate is treated with ten cubic centimeters of a ten per cent ammonium phosphate solution and again almost neutralized as described above, twenty-five cubic centimeters of the ammonium acetate solution added and warmed to 70°.
The precipitate obtained is once more dissolved and precipitated as above described, and is then collected upon a filter, washed, ignited, and weighed. The residue after ignition is dissolved in the crucible by heating with a little concentrated hydrochloric acid, and washed into a beaker. Any silicic acid present is separated by filtration, ignited, and weighed, and subtracted from the total weight of the precipitate. To the filtrate is added ammonia to diminish the acidity, but not sufficient to produce a precipitate and the clear solution is treated with thirty cubic centimeters of the ordinary ammoniacal citrate solution and fifteen cubic centimeters of magnesium mixture, and the [Pg 27] precipitation of the ammonium magnesium phosphate hastened by stirring with a glass rod.
It is advisable to always make the filtrate from the third precipitation slightly ammoniacal and to boil it for a long time. If the operation have been carried on correctly, there occurs only a slight precipitate of Ca₃P₂O₈ amounting only to a few milligrams. In some cases it may be necessary to dissolve the precipitate and reprecipitate the iron and aluminum phosphates a fourth time.
The whole time required for the triple precipitation, according to Hess, if all the operations be properly conducted, is from three to four hours. It is therefore possible by this variation of the acetate method to secure a determination of the iron and alumina as phosphates in the same time which is occupied by the Glaser-Jones method when the separation of lime is taken into account.
If the solution of the mineral phosphate employed contain any notable quantity of organic material, it must be destroyed by boiling with bromin or some other oxidation agent, before the precipitation by the acetate method is commenced.
The presence of silicic acid need not be taken into special consideration since this can be detected and determined in the phosphate precipitates after they have been ignited and weighed. While the determinations of the phosphoric acid in Hess’ method were made by precipitation in the presence of citrate, he found that they agree perfectly with the previous precipitations with molybdic solution.
35. Method of Glaser.—The principle on which this method rests depends on the preliminary removal of the lime by conversion into calcium sulfate and its precipitation in the presence of strong alcohol.[25] It is conducted as follows:
Five grams of the phosphate are dissolved in a mixture of twenty-five cubic centimeters of nitric acid of 1.2 specific gravity and about 12.5 cubic centimeters of hydrochloric acid of 1.12 specific gravity, and made up to a volume of half a liter, and filtered. One hundred cubic centimeters of the filtrate, equivalent to one gram of the substance, are placed in a quarter liter flask and twenty-five cubic centimeters [Pg 28] of sulfuric acid of 1.84 specific gravity added. The flask is allowed to stand for about five minutes and meanwhile shaken a few times. About 100 cubic centimeters of alcohol of ninety-five per cent are then added and the flask filled with alcohol to the mark and well shaken. A certain degree of concentration takes place and this is compensated for by lifting the stopper and filling again with alcohol to the mark and shaking a second time. After allowing to stand for half an hour the contents of the flask are filtered, 100 cubic centimeters of the filtrate being equal to four-tenths gram of the substance. This volume, filtered, is evaporated in a platinum dish until the alcohol is driven off. The alcohol-free residue is heated to boiling in a beaker with about fifty cubic centimeters of water. Ammonia is added to alkaline reaction, but in order to avoid strong effervescence it is not added during the boiling. The excess of ammonia is evaporated, the flask allowed to cool, the contents filtered, precipitate and filter washed with warm water, ignited, and the phosphates of iron and alumina weighed. Half of the weight of the precipitate represents the weight of Fe₂O₃ + Al₂O₃. The estimation, as before indicated, should be carried on without delay, the whole time required not exceeding from one and a half to two hours.
36. Jones’ Variation.—The method of Glaser described above, as practiced by the German chemists, has been found by Jones to be inaccurate on account of the alcohol not being added in sufficient quantity in the precipitation of calcium sulfate and for the additional reason that the amount of sulfuric acid added is more than is actually necessary[26]. Jones modifies the method as follows: Ten grams of the material are dissolved in nitro-hydrochloric acid and the solution made up to 500 cubic centimeters and filtered. Fifty cubic centimeters of this solution, representing one gram, are evaporated to twenty-five cubic centimeters and, while still hot, ten cubic centimeters of dilute sulfuric acid (one to five) added. The mixture is then well stirred and cooled. One hundred and fifty cubic centimeters of ninety-five per cent alcohol are next added and after stirring, the solution is allowed to stand three hours. The calcium sulfate is collected on a filter, washed with alcohol, and the filtrate and washings collected in an erlenmeyer. [Pg 29] The washing is completed when the last ten drops, after dilution with an equal volume of water, are not colored with a drop of methyl orange.
The moist calcium sulfate is transferred to a platinum crucible, the filter placed on it, the alcohol burned off, the filter incinerated, and the calcium sulfate ignited and weighed. The contents of the flask are heated to expel the alcohol, the residue washed into a beaker, made slightly alkaline with ammonia, and again heated till all the ammonia is driven off. This treatment is necessary to prevent the precipitate from being contaminated with magnesia. The precipitate is collected on a filter, washed four times with hot water, or water containing ammonium nitrate, dried, ignited, and weighed. One-half of the weight of the precipitate represents the weight of the ferric and aluminic oxids.
37. Estimation of Iron and Alumina in Phosphates by Crispo’s Method.—The phosphate of ferric iron is subject to a slight decomposition in presence of both hot and cold water with a tendency to the production of basic compounds. It is soluble to a slight extent in hot and cold acetic acid, almost insoluble in ammonium acetate, and quite insoluble in ammonium chlorid and nitrate. Aluminum phosphate is likewise soluble, to a slight degree, in acetic acid and ammonium acetate, and insoluble in ammonium chlorid and nitrate. The method of Crispo for the separation of iron and alumina in phosphates is based on the above properties.[27] Five grams of the mineral phosphate are dissolved in fifty cubic centimeters of aqua regia, composed of forty cubic centimeters of hydrochloric acid of 1.10, and ten of nitric acid of 1.20 specific gravity, and this solution is diluted to half a liter. To fifty cubic centimeters of the filtered solution are added two of ammonia (0.96) and fifty of a half saturated solution of ammonium chlorid, and the whole boiled. The liquid should remain clear, but if it become cloudy add a little dilute nitric acid, drop by drop, until the turbidity is removed, and then ten cubic centimeters of a saturated solution of ammonium acetate, and boil for three minutes, cool, and filter. The precipitate is washed twice with a ten per cent solution of ammonium chlorid and redissolved with two cubic centimeters of nitric acid, and [Pg 30] the filter washed with hot water. The phosphoric acid is separated by forty cubic centimeters of molybdate solution, and the precipitate washed three or four times with a one per cent nitric acid solution.
To the filtrate are added fifty cubic centimeters of a one-half saturated ammonium chlorid solution, ammonia is added in slight excess to produce precipitation and the mixture boiled for a few minutes. After filtering, the precipitate is washed with hot water three or four times, dissolved in two cubic centimeters of nitric acid, and the filter washed with hot water. Again, fifty cubic centimeters of half saturated ammonium chlorid are added and the precipitate thrown down once more by ammonia in slight excess. The precipitate is washed with hot water and finally ignited and weighed as iron and aluminum oxids.
According to Crispo, the original Glaser method, with its various modifications, is not to be considered reliable, and the choice lies between the molybdic method as usually practiced, and his own for the accurate estimation of iron and alumina. Manganese disturbs the accuracy of the results unless the directions given are carefully followed. Manganese phosphate is soluble at all temperatures below fifty. If then the mixture of the phosphates be allowed to cool before filtering, the iron and aluminum salts are not contaminated with manganese. This method of Crispo is somewhat tedious, but it is claimed that these variations of the molybdic method render it exact in respect of the determination of iron and alumina.
38. Method Employed in Geological Survey.—Chatard gives the following directions for conducting the Glaser-Jones process[28]: The distillation flask containing the alcoholic filtrate is connected with its condenser and heated on a water-bath until no more alcohol comes over. This distillate, if mixed with a little sodium carbonate and redistilled over quicklime, can be used over and over again, so that the expense for alcohol is really very slight, while in the use of the Glaser method, with its large amount of sulfuric acid, all the alcohol is lost.
When the distillation is ended the residue in the flask is washed into a platinum dish and evaporated to a small bulk on the water-bath. The dark brown color produced is due to the presence of organic matter and [Pg 31] this must be destroyed, as it prevents the complete precipitation of the phosphate in the subsequent operation.
The organic matter is best destroyed by removing the dish from the bath, adding a small quantity of pure sodium nitrate, and heating very carefully over the naked flame, keeping the dish well covered with a watch-glass to avoid spattering. The mass fuses to a colorless, viscous liquid, becoming glassy when cooled and is readily soluble in a hot very dilute solution of nitric acid. The solution transferred to a beaker is made distinctly alkaline with ammonia and carefully neutralized with acetic acid, diluted with hot water, boiled, and the precipitate allowed to settle, after which it is separated by filtration.
After the precipitate has been completely transferred to the filter, the washing is completed with a dilute solution of ammonium nitrate. The precipitate is dried, ignited, cooled, and weighed.
The determinations should be made in pairs, one portion being used for the estimation of the phosphoric acid by fusing with a little sodium carbonate, and the other, after fusion with sodium carbonate, is dissolved with sulfuric acid and the iron reduced and titrated with potassium permanganate solution. The filtrate from the iron and alumina determination is evaporated to a small bulk, made strongly ammoniacal and allowed to stand for some time when the magnesia present separates as ammonium magnesium phosphate which is determined in the usual way.
If, during the evaporation of the filtrate, any flocculent matter separate, it should be removed by filtration and examined before precipitating the magnesia.
39. Variation of Marioni and Fasselli.—Glaser’s method has been shown to be subject to errors by Marioni and Fasselli[29] in the following respects:
1. The precipitation of a small quantity of calcium phosphate with the ferric and aluminum phosphates.
2. The possible precipitation of basic phosphates if all the iron and alumina are not combined with phosphoric acid in the mineral.
3. The partial solubility of ferric and aluminum phosphates in dilute acetic acid. [Pg 32]
4. The decomposition of ferric orthophosphate into soluble acid phosphate and insoluble basic salt by boiling.
To avoid these errors the following procedure is proposed: From one to five grams of the phosphate are boiled in a flask for ten minutes with fifteen cubic centimeters of strong hydrochloric acid, and afterwards diluted with a double volume of water. A few crystals of potassium chlorate are added, and several drops of nitric acid, and the liquid boiled to expel chlorin. It is then filtered and washed until the volume of the filtered liquid amounts to 150 cubic centimeters. After cooling, a half gram of ammonium phosphate in solution is added, and two cubic centimeters of glacial acetic acid, followed by dilute ammonia, drop by drop, until a slight precipitate persists on stirring. Again the same quantity of acetic acid is added as above, well shaken, and left for two hours. The precipitate is collected on a filter and washed with a one per cent ammonium phosphate solution. The precipitate is dissolved by a minimum quantity of hydrochloric acid and the solution collected in the same vessel in which the precipitation took place. A second precipitation is conducted just as described above. The precipitate is washed as above described and ignited at a dull red heat. Half the weight obtained represents the ferric oxid and alumina.
40. Method of Ogilvie.—For the separation of alumina from phosphoric acid Ogilvie recommends that the filtrate from the phosphomolybdate precipitate be neutralized with ammonia, the precipitate thus formed redissolved in nitric acid, again precipitated with ammonia, filtered, ignited, and weighed as aluminum oxid.[30] If iron be present it will, of course, appear in the product. For use in the examination of mineral phosphates the method can not have a wide application without amendment.
41. Method of Krug and McElroy.—Krug and McElroy show that when sufficient alcohol is added to precipitate all of the calcium sulfate in the Glaser method, it will also cause a precipitation of a considerable quantity of iron, by means of which the calcium sulfate will be colored.[31] The presence of potassium and ammonium salts also affects very notably the precipitation of calcium. The method employed by them, in order to avoid these sources of error, is as follows: [Pg 33]
One hundred cubic centimeters, equivalent to one gram of the substance, in a nitric acid solution, are placed in a half liter flask and a solution of ammonium molybdate added until all the phosphoric acid has been precipitated. The addition of ammonium nitrate will hasten the separation of the ammonium phosphomolybdate. The liquid should be allowed to stand for twelve hours. The flask is then filled to the mark, the contents well shaken, filtered through a dry filter, and duplicate samples of 200 cubic centimeters each of the filtrate taken for analysis.
A small quantity of ammonium nitrate is dissolved in the liquid, and ammonia cautiously added, keeping the solution as cool as possible. The iron and alumina are precipitated as hydroxids. The mixed hydroxids are collected on a filter, washed with water, the filtrate and washings being collected in a beaker. The precipitate should be dissolved with a small quantity of a solution of ammonium nitrate and nitric acid, again precipitated with ammonia, filtered, washed, ignited, and weighed. This treatment is for the purpose of excluding all possibility of error from the presence of molybdic anhydrid. After weighing, the mixed oxids should be fused with sodium bisulfate, the magma dissolved in water, and the iron determined volumetrically with potassium permanganate after reduction to the ferrous state.
McElroy has further shown by experiments in this laboratory that even the molybdate method of separating the iron and alumina from phosphoric acid with the improvements as first suggested by Krug and himself, may not always give reliable results.[32] In a solution containing ferrous iron equivalent to 56.4 milligrams of ferric oxid, were placed enough of a solution of sodium phosphate to correspond to 100 milligrams of phosphorus pentoxid. The precipitate was dissolved by adding nitric acid, oxidized with bromin water, and the phosphoric acid thrown out with ammonium molybdate. The precipitate was washed with weak nitric acid and the combined filtrate and washings neutralized with ammonia. The resultant precipitate was dissolved in a solution of ammonium nitrate and nitric acid, filtered, and again precipitated with ammonia. In two instances the quantities of material recovered after ignition were 56.9 and 57.3 milligrams, respectively, instead of the theoretical amount, viz., 56.4 milligrams. [Pg 34]
When the work was repeated after the addition of 400 milligrams of calcium oxid the weight of the precipitate recovered was 62.3 and 63.1 milligrams in duplicate determinations. Similar determinations were made with a known weight, viz., 35.6 milligrams of alumina. The treatment of the mixture was precisely as indicated above for iron. The quantity of alumina finally obtained was 28.9 and 29.3 milligrams, respectively, in duplicate determinations. When the lime was added, however, the weights of alumina, recovered, fell to 19.8 and 20.6 milligrams, respectively. These results show that the molybdate method for the separation of iron and alumina in the presence of a large excess of lime and phosphoric acid is subject to widely varying results, but that the error due to the excess of iron in the weighed product is partly corrected by the one due to deficiency of alumina.
42. Method of Wyatt.—A method largely used in this country, both in private laboratories and by fertilizer factories, for determining iron and alumina, is described by Wyatt[33]. It is claimed for this method that, while it may not be strictly accurate, yet it is rapid and easy, and in the hands of trained analysts yields concordant results. Fifty cubic centimeters of the first solution of the sample in aqua regia, or an amount thereof equivalent to one gram of the phosphate, in a beaker, are rendered alkaline by ammonia. The resulting precipitate is first redissolved by hydrochloric acid, and a slight alkalinity is again produced with ammonia. Fifty cubic centimeters of strong acetic acid are next added, the mixture stirred and placed in a cool place and left until cold. The precipitate is then separated by filtration and washed twice with boiling water. The vessel holding the filtrate is replaced by the beaker in which the precipitation was made. The precipitate is dissolved in a little fifty per cent hot hydrochloric acid and the filter washed with hot water. After rendering slightly alkaline, as in the first instance, the treatment with acetic acid is repeated as described. The precipitate is washed this time, twice with cold water containing a little acetic acid and three times with hot water. The precipitate is dried, ignited, and weighed as iron and aluminum phosphate. Half of this weight may be taken to represent the quantity of iron and aluminum oxids. [Pg 35]
To separate the iron and alumina the precipitate just described is dissolved in hot hydrochloric acid, filtered into a 100 cubic centimeter flask, and made up to the mark by hot wash-water.
The phosphoric acid is determined in one-half of the filtrate and in the remaining half the iron is reduced with zinc and determined with potassium permanganate in the usual way. The phosphoric acid and iron having been thus determined the alumina is estimated by difference. The chief objection to this process is in the excessive quantity of acetic acid used and the danger of solution of the precipitated phosphates caused thereby.
43. Estimation of the Lime and Magnesia.—The filtrate and washings from the first precipitation, (paragraph 41,) of iron and alumina in the method of Krug and McElroy, above described, are collected and sufficient ammonium oxalate is added to precipitate the calcium. The precipitated calcium is very fine and should be collected on a gooch, under pressure. The filtrate and washings from the calcium precipitate are again collected, and a solution of sodium phosphate added to precipitate the magnesia. The solution must be kept cool and slightly alkaline with ammonia during the above operations in order to prevent the separation of molybdic anhydrid.
44. Estimation of Sulfuric Acid.—As a rule, sulfates are not abundant in mineral phosphates. In case the samples are pyritiferous, however, considerable quantities of sulfuric acid may be found after treatment with aqua regia.
The acid is precipitated with barium chlorid, in the usual way, in an aliquot portion of the filtrate first obtained. The precipitate of barium sulfate is washed with hot water until clean, dried, ignited, and weighed. If the portion of the filtrate taken represent half a gram of the original material then the weight of barium sulfate obtained multiplied by 0.6858 will give the quantity of sulfur trioxid in one gram.
45. Estimation of Fluorin by the Method of Berzelius as Modified by Chatard.—The method of estimating fluorin as proposed by Berzelius, has been found quite satisfactory in the laboratory of the [Pg 36] Geological Survey, with the modifications given below.[34]
Two grams of the phosphate are intimately mixed in a large platinum crucible with three grams of precipitated silica and twelve grams of pure sodium carbonate, and the mixture is gradually brought to clear fusion over the blast-lamp. When the fusion is complete the melt is spread over the walls of the crucible, which is then rapidly cooled (preferably by a blast of air). If this have been properly done, the mass separates easily from the crucible, and the subsequent leaching is hastened. The mass, detached from the crucible, is put into a platinum dish into which whatever remains adhering to the crucible, or its lid, is also washed with hot water. A reasonable amount of hot water is now put into the dish, which is covered and digested on the water-bath until the mass is thoroughly disintegrated. To hasten this, the supernatant liquid may, after awhile, be poured off, the residue being washed into a small porcelain mortar, ground up, returned to the dish and boiled with fresh water until no hard grains are left. The total liquid is then filtered, and the residue is washed with hot water. The filtrate (which should amount to about half a liter) is nearly neutralized with nitric acid (methyl orange being used as indicator), some pure sodium bicarbonate is at once added, and the solution (in a platinum dish, if one large enough is at disposal, otherwise in a beaker) is placed on the water-bath, when it speedily becomes turbid through separation of silica. As soon as the solution is warm it is removed from the bath, stirred, allowed to stand for two or three hours, and then filtered by means of the filter-pump and washed with cold water.
The filtrate is concentrated to about a quarter of a liter and nearly neutralized, as before, some sodium carbonate is added, and the phosphoric acid is precipitated with silver nitrate in excess. The precipitate is separated by filtration and washed with hot water, and the excess of silver in the filtrate is removed with sodium chlorid.
The filtrate from the silver chlorid (after addition of some sodium bicarbonate) is evaporated to its crystallizing point, then cooled and diluted with cold water; still more sodium bicarbonate is added, and the whole is allowed to stand, when additional silica will separate, and this is to be removed by filtration. [Pg 37]
This final solution is nearly neutralized, as before; a little sodium carbonate solution is added; it is heated to boiling and an excess of solution of calcium chlorid is added. The precipitate of calcium fluorid and carbonate must be boiled for a few minutes, when it can be easily filtered and washed with hot water. The precipitate is then washed from the filter into a small platinum dish and evaporated to dryness, while the filter, after being partially dried and used to wipe off any particles of the precipitate adhering to the dish in which it was formed, is burned, and the ash is added to the main precipitate. This, when dry, is ignited, and allowed to cool; dilute acetic acid is added in excess, and the whole is evaporated to dryness, being kept on the water-bath until all odor of acetic acid has disappeared. The residue is then treated with hot water, digested, filtered on a small filter, washed with hot water, partially dried, put into a crucible, carefully ignited, and weighed as calcium fluorid. The calcium fluorid is then dissolved in sulfuric acid by gentle heating and agitation, evaporated to dryness on a radiator, ignited at full red heat, and weighed as calcium sulfate. From this weight the equivalent weight of calcium fluorid should be calculated, and this should be very close to that actually found as above, but should never exceed it. The difference, which is generally about a milligram (sometimes more), is due to silica precipitated with the fluorid. The percentage of fluorin is, therefore, always calculated from the weight of the sulfate, and not from that of the fluorid obtained.
The main improvements in this method are the use of sodium bicarbonate to separate the silica, and the keeping of the earlier solutions as dilute as possible, which can not be done, if ammonium carbonate be used for the separation of the silica. These changes make the fluorin estimation, although still tedious, far more rapid than before, and the results are very satisfactory.
46. Modification of Wyatt.—By reason of the tediousness of the method of Chatard given above, Wyatt has sought to shorten the process by the following modification:[35]
Five grams of the finely ground phosphate are fused in a platinum dish with fifteen grams of the mixed carbonates of sodium and potassium and three grams of fine sand. After fusing very thoroughly with a strong [Pg 38] heat for a quarter of an hour, the dish is removed from the fire and cooled. Its contents, dissolved in hot water, are then put into a half liter flask, and a considerable excess of ammonium carbonate is added to the liquid. All the soluble silica falls out of solution, and the flask, after cooling, is made up to the mark with distilled water, well shaken, and then set aside for twenty-four hours to settle. At the end of this time 200 cubic centimeters are carefully decanted through a filter; the filter is well washed, and the filtrate, after being nearly neutralized with hydrochloric acid, is treated with an excess of calcium chlorid solution.
The precipitate, consisting of phosphate, fluorid, and some calcium carbonate, is allowed to settle, and is then carefully washed with boiling water, first by decantation several times, and finally on the filter. After being properly dried in the gas-oven, calcined, and cooled, the residue is treated with acetic acid, placed upon the water-bath, and evaporated to complete dryness.
The calcium acetate is now well washed out by several treatments with boiling water, and the residue is brought upon a filter, dried, calcined, and weighed. The weight represents the calcium phosphate and fluorid contained in two grams of the original sample; and if the calcium phosphate in the residue be determined, according to the usual methods, the difference will be calcium fluorid and may be thus estimated.
Example.—Assuming the calcined residue of calcium phosphate and fluorid in two grains of the original sample to have amounted to one and six-tenths gram and the calcium phosphate in this quantity to have been determined as 1.540 gram, the calcium fluorid is thus proved to be 0.060 gram, and, therefore, 2: 0.60::100: x = 3 per cent calcium fluorid which, multiplied by 0.4897, gives 1.46 per cent of fluorin.
The above method, while shorter, is not to be preferred to the Chatard process when great accuracy is desired. All the soluble silica may not fall out of the solution as Wyatt says. Finally the fluorin is calculated from small differences in the weight of very heavy precipitates and all the error of the process may be found affecting the numbers for fluorin. For commercial purposes, however, the method is to be recommended for its comparative brevity. [Pg 39]
47. Preliminary Considerations.—The chief sources of the phosphoric acid in commercial fertilizers are the mineral phosphates and bones. In respect of the analyses of mineral phosphates detailed directions have been given in the preceding pages. Bones are valuable for fertilizing materials, both because of their content of phosphoric acid and of their organic nitrogen. The methods of treating bones for their phosphoric acid will be found in the general methods for fertilizing materials, and their nitrogen content can be determined by the processes to be described hereafter. Other fertilizing materials also contain phosphorus, as ashes, tankage, oil cakes, and other organic products. In general, the methods for determining the phosphoric acid is the same in all cases, but the means of destroying the organic matter precedent to the analysis vary in different cases. In most cases a simple ignition is sufficient, while, if the phosphorus be found in certain organic products, the oxidation must be accomplished by one of the methods described in the processes adopted by the official chemists, or by the means described in volume first, paragraph 378 or 382. In all cases of acid phosphates and superphosphates, the water and ammonium citrate soluble phosphoric acid is to be determined as well as the total. In basic slags the amount soluble in ammonium citrate or dilute citric acid is also to be ascertained.
In all cases where soluble or so-called reverted acid is to be considered, the analysis must be performed without previous desiccation or ignition. If water content or loss on ignition are to be considered, the operation to determine them must be conducted on a separate part of the sample.
The methods of analysis which have been adopted by associations of chemists should be given the preference in the conduct of the work, although it must be admitted that they may contain sources of error, and may be in no respect superior to processes employed by chemists in their private capacity. In this country the methods adopted by the Association of Official Agricultural Chemists should be followed [Pg 40] as closely as possible. The great merit of other methods, however, must not be denied. Especially those methods which shorten the time required or diminish the labor and expense of the analysis are worthy of careful consideration. In factory work, for instance, it is often far more important for the chemist to be able to rapidly determine the phosphoric acid in a great number of samples with approximate accuracy than to confine his work to one with absolute precision. Some of the shorter methods, moreover, notably the citrate process, appear to be quite, if not altogether, as reliable as the molybdate method, while in the case of the uranium volumetric process, it must not be forgotten that it is almost the only one practiced in France. Other volumetric processes are given in full, as, for instance, the one perfected by Pemberton, but data are still lacking to justify their strong recommendation. It should be remembered that this manual is not written for the beginner but rather for the chemist already acquainted with the principles and practice of general chemical analysis, and it is, therefore, expected that each analyst will make intelligent use of the data placed at his disposal.
48. Determination of Phosphoric Acid with Preliminary Precipitation as Stannic Phosphate.—This method, once much in use and highly recommended, is now almost unknown among the processes of fertilizer control. It was first proposed and described by Girard, and rests on the precipitation of the phosphoric acid in a nitric acid solution by means of metallic tin.[36] The stannic acid formed by the oxidation of the tin unites with the phosphoric acid held in a free state by the nitric acid. The precipitation of the phosphoric acid is said to be complete, but a trace of it has been found in the iron and alumina subsequently separated from the solution. The precipitate obtained is dissolved in caustic potash, whereby soluble potassium metastannate and phosphate are obtained. Following is the method of conducting the analysis as described by Crookes:[37]
The phosphate should be dissolved in nitric acid, and any chlorin present be expelled by repeated evaporations with the solvent. Finally, to the evaporated mass the strongest nitric acid is added. Pure [Pg 41] tin-foil is added and heat applied. The phosphoric acid is precipitated by the stannic acid formed. The quantity of tin used should be from six to eight times as great as that of the phosphoric acid present.
The precipitate is collected on a filter, washed, and dissolved in caustic potash. The solution is saturated with hydrogen sulfid, and on adding acetic acid in slight excess the tin sulfid is separated and removed by filtration. The whole of the phosphoric acid, supposed to be almost free of tin, is now found in the filtrate. The filtrate is concentrated to small bulk and any tin sulfid present separated by filtering, and the phosphoric acid finally removed from the ammoniacal filtrate by precipitation with magnesia mixture. The chief difficulties of this method are to be found, on the one hand, in the retention of some of the phosphoric acid by the iron and alumina which may be present, and on the other, in the presence of some tin in the final magnesium pyrophosphate. If the tin be all removed as sulfid, the latter source of error will be avoided. It is difficult to secure pure metallic tin, and this is another disturbing element in the process. It can not be recommended for the work which agricultural analysts are usually called on to perform.[38]
49. Water-Soluble Phosphoric Acid.—The method of procedure recommended by the Association of Official Chemists is as follows:[39] Place two grams of the sample in a nine centimeter filter; wash with successive small portions of cold water, allowing each portion to pass through before adding more, until the filtrate measures about 250 cubic centimeters. If the filtrate be turbid, add a little nitric acid. Make up to any convenient definite volume; mix well; take any convenient portion and proceed as under total phosphoric acid.
50. Citrate-Insoluble Phosphoric Acid.—Heat 100 cubic centimeters of strictly neutral ammonium citrate solution of 1.09 specific gravity to 65° in a flask placed in a bath of warm water, keeping the flask loosely stoppered to prevent evaporation. When the citrate solution in the flask has reached 65°, drop into it the filter containing the washed residue from the water-soluble phosphoric acid determination, close tightly with a smooth rubber stopper, and shake violently until the filter paper is reduced to a pulp. Place the flask [Pg 42] back into the bath and maintain the water in the bath at such a temperature that the contents of the flask will stand at exactly 65°. Shake the flask every five minutes. At the expiration of exactly thirty minutes from the time the filter and residue were introduced, remove the flask from the bath and immediately filter as rapidly as possible. It has been shown by Sanborn, in this laboratory, that the filtration is greatly facilitated by adding asbestos pulp. Wash thoroughly with water at 65°. Transfer the filter and its contents to a crucible, ignite until all organic matter is destroyed, add from ten to fifteen cubic centimeters of strong hydrochloric acid, and digest until all phosphate is dissolved; or return the filter with contents to the digestion flask, add from thirty to thirty-five cubic centimeters of strong nitric, and from five to ten cubic centimeters of strong hydrochloric acid, and boil until all the phosphate is dissolved. Dilute the solution to 200 cubic centimeters. If desired, the filter and its contents can be treated according to methods (1), (2), or (3), under total phosphoric acid. Mix well; filter through a dry filter; take a definite portion of the filtrate and proceed as under total phosphoric acid.
In case a determination of citrate-insoluble phosphoric acid be required in non-acidulated goods it is to be made by treating two grams of the phosphatic material, without previous washing with water, precisely in the way above described, except that in case the substance contain much animal matter (bone, fish, etc.), the residue insoluble in ammonium citrate is to be treated by one of the processes described below under total phosphoric acid, (1), (2), or (3).
51. Total Phosphoric Acid.—In case of ignition the residual material is to be dissolved in hydrochloric acid. The following methods of treating the raw material, using two grams in each case, may be employed: (1) Evaporate with five cubic centimeters of magnesium nitrate, ignite, and dissolve in hydrochloric acid. (2) Boil in a Kjeldahl flask graduated to 250 cubic centimeters, with from twenty to thirty cubic centimeters of strong sulfuric acid, adding from two to four grams of sodium or potassium nitrate at the beginning of the digestion and a small quantity after the solution has become nearly colorless; or adding the nitrate in small portions from time to time. [Pg 43] After the solution is colorless, add 150 cubic centimeters of water and boil for a few minutes, cool, and make up to volume. (3) Digest with strong sulfuric acid and such other reagents as are used in either the plain or modified Kjeldahl or Gunning methods for estimating nitrogen. Do not add any potassium permanganate, but after the solution has become colorless add about 100 cubic centimeters of water and boil for a few minutes, cool, and make up to a convenient volume; two and five-tenths grams of substance and a digestion flask graduated to 250 cubic centimeters are recommended. This method will be found convenient when both the nitrogen and the total phosphoric acid are to be determined in a fertilizer. In this case, after diluting the volume and mixing, a part for the estimation of nitrogen, may be removed with a pipette and the remainder then filtered through a dry filter and a portion taken for the determination of the total phosphoric acid. (4) Dissolve in thirty cubic centimeters of concentrated nitric acid and a small quantity of hydrochloric acid. (5) Add thirty cubic centimeters of concentrated hydrochloric acid, heat, and add cautiously, in small quantities at a time, about five-tenths gram of finely-pulverized potassium chlorate. (6) Dissolve in from fifteen to thirty cubic centimeters of strong hydrochloric and from three to ten cubic centimeters of nitric acid. This method is recommended for fertilizers containing much iron or aluminum phosphate. Boil until all phosphates are dissolved and all organic matter is destroyed; cool and dilute to 200 or 250 cubic centimeters; mix and pass through a dry filter; take an aliquot part of the filtrate corresponding to a quarter, half, or one gram, neutralize with ammonia, and clear with a few drops of nitric acid. In case hydrochloric or sulfuric acid have been used as a solvent, add about fifteen grams of dry ammonium nitrate.
To the hot solutions, for every decigram of phosphorus pentoxid that is present, add fifty cubic centimeters of molybdic solution. Digest at about 65° for an hour, filter, and wash with water or ammonium nitrate solution. Test the filtrate by renewed digestion and the addition of more molybdic solution. Dissolve the precipitate on the filter with ammonia and hot water and wash into a beaker to a bulk of not more than [Pg 44] 100 cubic centimeters. Nearly neutralize with hydrochloric acid, cool, and add magnesia mixture from a burette; add slowly (about one drop per second), stirring vigorously. After fifteen minutes add thirty cubic centimeters of ammonia solution of 0.95 density. Let stand for some time; two hours are usually enough. Filter, wash with dilute ammonia, ignite gently at first and then at white heat for ten minutes, and weigh. For the quantity of magnesia mixture to be added see paragraph 21.
52. Citrate-Soluble Phosphoric Acid.—The sum of the water-soluble and citrate-insoluble subtracted from the total gives the citrate-soluble phosphoric acid.
53. Preparation of Reagents.—(1) Ammonium Citrate Solution.—(a) Mix 370 grams of commercial citric acid with 1,500 cubic centimeters of water, nearly neutralize with commercial ammonia, cool, add ammonia until exactly neutral (testing with saturated alcoholic solution of corallin) and bring to a volume of two liters. Test the specific gravity, which should be 1.09 at 20°, before using.
(b) Alternate Method.—To 370 grams of commercial citric acid add commercial ammonia, of 0.96 specific gravity, until nearly neutral; reduce the specific gravity to nearly 1.09 and proceed as follows: Prepare a solution of fused calcium chlorid 200 grams to the liter, and add four volumes of strong alcohol. Make the mixture exactly neutral, using a small amount of freshly prepared corallin solution as a preliminary indicator, and test finally by withdrawing a portion, diluting with an equal volume of water, and testing with cochineal solution. Fifty cubic centimeters of this solution will precipitate the citric acid from ten cubic centimeters of the citrate solution. To ten cubic centimeters of the nearly neutral citrate solution add fifty cubic centimeters of the alcoholic calcium chlorid solution, stir well, filter at once through a folded filter, dilute with an equal volume of water, and test the reaction with neutral solution of cochineal. If acid or alkaline, add ammonia or citric acid, as the case may be, to the citrate solution, mix, and test again as before. Repeat this process until a neutral reaction of the citrate solution is obtained. At the end the specific gravity must be 1.09 at 20°. [Pg 45]
(2) Molybdic Solution.—See paragraph 22.
(3) Ammonium Nitrate Solution.—Dissolve 200 grams of commercial ammonium nitrate in water and bring to a volume of two liters.
(4) Magnesia Mixture.—See paragraph 22.
(5) Dilute Ammonia for Washing.—See paragraph 22.
(6) Magnesium Nitrate.—Dissolve 320 grams of calcined magnesia in nitric acid, avoiding an excess of the latter; then add a little calcined magnesia in excess, boil, filter from the excess of magnesia, ferric oxid, etc., and bring to volume of two liters.
54. Official Methods with Norwegian Fertilizers.—The Director of the Chemical Control Station of Norway, expresses the opinion, that for Norwegian, Swedish, Danish, and German conditions, the American methods for the determination of phosphoric acid, notwithstanding their analytical exactness, are quite inapplicable.[40] In those countries are found many, in part, poorly pulverized and badly mixed manures, such as ammonium superphosphate, potassium superphosphate, and potassium ammonium superphosphate, and these can not usually be so well pulverized and mixed that one can take out a true average sample of from two to two and five-tenths grams. Care in the analysis is useless when the material employed does not represent the average condition of the materials investigated. Therefore, in the countries named, often from ten to twenty grams, and almost never less than five grams of substance are taken in the preparation of the solutions, except for instance, in the determination of nitrogen and reverted phosphoric acid.
55. The Molybdic Acid Method, as Practiced by Direction of the Union of the German Experiment Stations.—The method adopted by the German Experiment Stations is essentially that used at Halle.[41] The samples are brought into solution in the following way: For the estimation of phosphoric acid in bone-meal, fish-guano and raw phosphates, and the total phosphoric acid in superphosphates, five grams of the sample are dissolved in fifty cubic centimeters of aqua regia, made of three parts of hydrochloric acid of 1.12 specific gravity and one part of nitric acid of 1.25 specific gravity, or the [Pg 46] solution may be made of a mixture of twenty cubic centimeters of nitric acid of 1.42 specific gravity and fifty cubic centimeters of sulfuric acid of 1.8 specific gravity. The boiling should continue for half an hour. The solution is made up to half a liter and filtered. Fifty cubic centimeters of the filtrate containing the phosphoric acid, with double superphosphates twenty-five cubic centimeters, are digested with 200 cubic centimeters of ammonium molybdate solution for three hours at 50° in a water-bath and, after cooling, filtered, so that as little as possible of the precipitate is collected upon the filter, which is made of strong paper.
The yellow precipitate is washed by decantation in the flask nine times with twenty cubic centimeters of molybdic solution diluted with one volume of water and the filter washed out once with the same quantity of liquid. The funnel, with the filter, is immediately placed upon the flask and the portion of the precipitate collected in the filter dissolved in five per cent ammonia, which is easily accomplished by throwing ammonia upon it from a wash-bottle. Afterwards the filter is washed with a sufficient quantity of hot water and finally removed. The contents of the flask are neutralized warm, with hydrochloric acid, the acid being added until the precipitate first formed, after continued shaking, is again dissolved in the liquid. The solution is then cooled and treated, drop by drop, with constant stirring, with twenty cubic centimeters of magnesia mixture. Finally twenty-five cubic centimeters of dilute ammonia solution are added, the precipitate is not shaken, and, after two hours, is filtered through a gooch.
For the filtering of the ammonium magnesium phosphate by the molybdic method, freshly prepared felts are always employed since the remarkably fine crystalline precipitates will pass through a filter which has once been used. It is necessary also that special precautions be taken in the ignition. The crucible should be heated in a platinum cap, which has the purpose of protecting the contents of the crucible from the access of reducing gases during the ignition. After redness has been reached the cap can be removed and the crucible transferred to a blast where it is strongly ignited for ten minutes before weighing. The precipitate should be pure white. [Pg 47]
The molybdic solution is prepared as follows: 150 grams of ammonium molybdate are dissolved in a liter of water, and after the solution is completely cooled, poured into a liter of nitric acid of 1.2 specific gravity.
56. Estimation of Soluble Phosphoric Acid.—1. The extraction of the superphosphates is made as follows: Twenty grams of the superphosphates are placed in a liter flask with 800 cubic centimeters of water and shaken continuously for thirty minutes. The flask is then filled with water to the mark and the whole again thoroughly shaken and filtered. For shaking, a machine is recommended, driven by hand or water power. The normal rate of the machine is fixed at 150 turns per minute.
2. The solution of the total superphosphates, obtained as above, must be boiled with nitric acid before the precipitation of the phosphoric acid in order to convert any phosphoric acid present as pyrophosphoric into tribasic phosphoric acid. For each twenty-five cubic centimeters of the superphosphate solution ten cubic centimeters of concentrated nitric acid are added and the mixture boiled.
3. The precipitation of the phosphoric acid is conducted by the molybdenum method as usually practiced.
4. For the estimation of iron and alumina in each of the superphosphates the Glaser method is recommended provisionally.
57. Methods for Phosphoric Acid used in the Norway Stations.[42]—1. Description of the Method for Total Phosphoric Acid.—For determining the phosphoric acid in bone-meal, fish-guano, and superphosphates, five grams of the substance, with twenty cubic centimeters of nitric acid of 1.42 specific gravity, and fifty cubic centimeters of sulfuric acid of 1.8 specific gravity, are boiled half an hour in a half liter flask, diluted with water, and after cooling, made up to the mark. Fifty cubic centimeters of the filtrate are made alkaline with ammonia, then acid with nitric acid, precipitated with fifty cubic centimeters of molybdic solution for every one-tenth gram of phosphorus pentoxid present, heated over the water-bath for one hour, and allowed to stand twelve hours more, when the supernatant liquid is separated by decantation the precipitate washed thoroughly with dilute molybdate solution (1: 4) dissolved in warm dilute ammonia, [Pg 48] and the filter washed with hot water. The ammoniacal solution is neutralized with hydrochloric acid, cooled, mixed, drop by drop, with constant stirring, with from ten to twenty cubic centimeters of magnesia mixture, and after a quarter of an hour one-third the volume of ten per cent ammonia is added. This is allowed to stand two hours, is filtered, washed with five per cent of ammonia until the disappearance of the chlorin reaction, dried, burned in an open crucible over a bunsen, and finally for a quarter of an hour, in a covered crucible heated over the blast.
2. Water-Soluble Phosphoric Acid.—To twenty grams of the substance in a liter flask, are added 800 cubic centimeters of water, and shaken every fifteen minutes for two hours; the volume made up to the mark and the phosphoric acid in fifty cubic centimeters of the filtrate, equaling one gram substance, is determined as under total.
3. Reverted Citrate-Soluble Phosphoric Acid.—Two and five-tenths grams substance are rubbed up with water, then washed upon the filter with about 100 cubic centimeters of water, the residue on the filter washed into a flask with a part of the measured citrate solution, and digested one hour at 35° to 40° with 200 cubic centimeters of Petermann’s citrate solution. The water and citrate extracts are made up to a quarter of a liter each, and the phosphoric acid determined in from twenty-five to fifty cubic centimeters, according to the quantity present.
Solutions. 1. Molybdate Solution.—375 grams of ammonium molybdate are dissolved in two and five-tenths liters of water, and the solution poured into two and five-tenths liters of nitric acid of 1.20 specific gravity.
2. Magnesia Mixture.—275 grams of crystallized magnesium chlorid and 350 grams of ammonium chlorid are dissolved in 3250 cubic centimeters of water and filled up to five liters with ammonia of 0.96 specific gravity.
3. Petermann’s Solution.—One kilogram of citric acid is dissolved in about two liters of water and 1350 cubic centimeters of ammonia of 0.925 specific gravity and filled up with water to 5750 cubic centimeters. The solution then has a specific gravity of 1.09; 300 cubic centimeters of ammonia of 0.925 specific gravity are now added. [Pg 49]
58. Swedish Official Method for Determination of Phosphoric Acid.[43]—The Swedish chemists determine phosphoric acid in fertilizers both by the molybdate and the citrate methods. These methods carefully conducted according to the directions given below, give very concordant results. In doubtful cases the former method is taken as the deciding one, it having proved by long practice to give very satisfactory results.
Reagents for the Molybdate Method.—1. Molybdic Solution.—Prepared by dissolving 100 grams of finely powdered molybdic acid with heat, in 400 grams of eight per cent ammonia of 0.967 specific gravity and pouring the solution into 1,500 grams of nitric acid of one and two-tenths specific gravity; or else by dissolving 150 grams ammonium molybdate in one liter of hot water, and pouring the solution into one liter of nitric acid of 1.2 specific gravity. Prepared in this way, the molybdic solution will contain, in the former case five per cent, in the latter case from five to six per cent of molybdic acid, and 100 cubic centimeters of it are required for precipitating one-tenth gram of phosphorus pentoxid.
2. Magnesia Mixture.—Prepared from 110 grams of crystallized magnesium chlorid, 140 grams ammonium chlorid, 700 grams of eight per cent ammonia of 0.967 specific gravity and 1,300 grams of distilled water. The mixture is filtered after a few days, if necessary; ten cubic centimeters of the same are required for precipitating one-tenth gram of phosphorus pentoxid.
3. Ten per cent ammonia of 0.959 specific gravity.
(a) Water-Soluble Phosphoric Acid.—1. Preparation of the Aqueous Solution.—Of superphosphates and in general fertilizers containing water-soluble phosphoric acid, a sample of twenty grams is taken, and water poured over it in a mortar; lumps are crushed lightly, but completely with the pestle without pulverizing it finer; the whole mass is then washed into a graduated flask holding one liter, which at once is filled up to the mark. The volume taken up by the residue insoluble in water, is left out of consideration in the calculation. The sample is left standing in the flask (which is occasionally shaken) at the ordinary temperature of the room for two hours, and the solution is then filtered. [Pg 50]
2. The Determination.—Take twenty-five cubic centimeters of the superphosphate solution thus prepared (when a twenty per cent sample is taken equal to one-tenth gram phosphorus pentoxid); add a quantity of molybdic solution sufficient for complete precipitation, leave standing for four hours in a beaker covered with a watch-glass; decant the solution through a small filter, wash the precipitate first by decantation, then on filter, with a mixture containing 100 parts molybdic solution, twenty parts nitric acid of 1.2 specific gravity, and eighty parts water, until a few drops put into alcohol, to which some dilute sulfuric acid has been added, does not, any longer, cause turbidity. The molybdic precipitate is now washed with but little water from the filter into a beaker, and particles adhering to the filter are dissolved by a hot mixture of one part ammonia and three parts water, which is allowed to flow into the beaker till the precipitate is, finally, completely dissolved in it. To the clear solution, add dilute hydrochloric acid while stirring, till the yellow precipitate formed by the acid is no longer immediately dissolved; then add from six to eight cubic centimeters of ammonia through the filter. The volume of the solution is not to exceed seventy-five cubic centimeters. It is now cooled completely and one cubic centimeter of magnesia mixture is added from a burette for every centigram of phosphorus pentoxid which it is expected to contain, and finally one-quarter of its volume of ammonia is added. The precipitate may be filtered after four hours. This is washed on the filter, preferably by means of suction, with a mixture of one part ammonia and three parts water till the filtrate is entirely free from chlorin. After drying, heat the precipitate, first gently, then stronger, and finally with a blast for a few minutes and then weigh it.
Treated with hydrochloric acid it must leave no insoluble residue (SiO₂), nor should hydrogen sulfid cause any precipitation in the solution thus formed (MoO₃).
(b) Total Phosphoric Acid.—1. In Superphosphates.—For the determination of total phosphoric acid, treat a weighed quantity of the superphosphate with nitric acid, if necessary to bring a difficultly soluble residue into solution, with addition of hydrochloric acid, or of potassium chlorate, to destroy organic matter [Pg 51] present. Dilute the solution to a definite volume, and determine the phosphoric acid in a measured quantity of the same, as directed under (a) 2; if hydrochloric acid or potassium chlorate, be applied in the preparation of the solution, however, not till the measured quantity has been repeatedly evaporated to dryness with concentrated nitric acid.
2. In Bone-meal.—Destroy organic matter in five grams of the sample by ignition, dissolve the residue in nitric acid, filter from the insoluble residue, dilute the filtrate to half a liter, take an aliquot part containing about one-tenth gram phosphorus pentoxid and determine the phosphoric acid as directed under (a) 2.
3. In Fish-guano (and other fertilizing materials of organic origin).—The organic matter cannot here be removed by simple ignition, as in this way a loss of phosphorus may take place; It is therefore destroyed either in the wet way through nitric acid and potassium chlorate or in the dry way by fusion with a mixture of potassium nitrate and sodium carbonate, otherwise the procedure is as in (b) 1.
4. In Mineral Phosphates.—Determine the phosphoric acid in a solution obtained by nitric acid; organic matter is destroyed preferably in the wet way.
5. In Basic Slag.—Dissolve ten grams of powdered slag by treating it with 100 cubic centimeters of fuming hydrochloric acid with heat; wash the solution into a graduated half liter flask, fill to the mark, shake well, and filter. Determine the phosphoric acid in twenty-five cubic centimeters of the clear filtrate, according to (a) 2, after having first, however, evaporated the solution to dryness and then at least three times evaporated the residue to dryness with concentrated nitric acid.
59. Method Employed by the Royal Experiment Station of Holland.—A. Soluble Phosphoric Acid.[44]—The necessary reagents are:
(1) Molybdate solution, made by dissolving 150 grams of ammonium molybdate in a liter of water and pouring the solution into a liter of nitric acid of 1.20 specific gravity.
(2) A ten per cent solution of ammonium nitrate.
(3) Strong and dilute ammonia, the latter being between two and five-tenths and three per cent of 0.988 specific gravity. [Pg 52]
(4) Magnesia mixture made by dissolving 110 grams of crystallized magnesium chlorid, 140 grams of ammonium chlorid, and 700 cubic centimeters of ammonia of 0.96 specific gravity in water and bringing the solution to two liters.
(5) Ammoniacal citrate solution, made by dissolving 500 grams of citric acid in a liter of water, and mixing with four liters of ten per cent ammonia of 0.96 specific gravity.
Manipulation.—Place twenty grams of substance in a mortar together with some cold distilled water or pure rain water, stir, and decant the water and suspended matters into a liter flask. After this has been repeated several times, rub up the residual mass and wash it all into the flask. Fill up to about 900 cubic centimeters and allow to stand two hours (twenty-four hours in the case of double phosphates with more than twenty-two per cent of soluble phosphoric acid), shaking repeatedly; or shaking continuously, for half an hour. Fill up to the liter mark and filter through a dry filter. Take portions of twenty-five or fifty cubic centimeters for each determination, add 100 cubic centimeters of molybdate solution for each 100 milligrams of phosphorus pentoxid present, warm to about 80° for an hour, filter, and wash the precipitate with the ammonium nitrate solution. Add a little molybdate solution to the filtrate, warm, and, if a fresh precipitate be observed, it is to be added to the first. The precipitate is to be dissolved in ammonia, and hydrochloric acid carefully added until the precipitate caused by it only slowly redissolves on stirring. The phosphoric acid is precipitated from the clear liquid which is still ammoniacal with magnesia mixture, using ten cubic centimeters for each 100 milligrams of phosphorus pentoxid present. This is added, drop by drop, and the liquid kept stirred during the addition. Allow it to stand at least two hours, filter, wash with dilute ammonia, dry, and ignite. This last is done at first with a very small flame but is finished with the blast-lamp or in a Rössler furnace. To insure burning to whiteness, nitric acid may be used, but not more than one or two drops.
B. Total Phosphoric Acid.—(1) For bone and flesh-meal, fish-guano, and similar fertilizers the reagents necessary are the same as before.
Carefully burn five grams to ash, boil the ash for half an hour with [Pg 53] nitric acid of 1.32 specific gravity, dilute with water, and, after cooling, dilute to 500 cubic centimeters. Filter through a dry filter and take fifty cubic centimeters of the filtrate. Add 100 cubic centimeters of the molybdate solution for each 100 milligrams of phosphorus pentoxid present. Treat further as before described.
(2) Phosphates, guanos, bone-black, etc.
One gram of substance, after powdering, and, if necessary, igniting, is covered with four cubic centimeters of hydrochloric acid of 1.13 specific gravity and a little water and heated for an hour and a half. Evaporate to dryness without filtration, making repeated additions of nitric acid until no more vapors of hydrochloric acid are evolved. Boil the residue with nitric acid, cool, make up to 100 cubic centimeters with water, and shake. Filter and treat fifty cubic centimeters of the resulting solution by the molybdate method and proceed further as before described.
60. Sources of Error in the Molybdate Method.—When conducted with proper care, the gravimetric molybdate method is one of the most exact processes known to analytical chemistry.
There are, however, some sources of error in the process which should be avoided as carefully as possible or taken into account.
1. Error Due to Occluded Silica.—When silica passes into solution in the original sample, and this may be the case especially with mineral phosphates, it may appear both in the yellow precipitate and in the final magnesium pyrophosphate. In all such cases the residue, after ignition, should be dissolved in hydrochloric acid, and any insoluble residue weighed as silica and deducted from the first weight. If the silica be removed by evaporating the solution of the original material to dryness, and igniting to destroy organic matter, care must be taken to reconvert all phosphoric acid into the ortho form by long boiling with nitric acid before precipitation.
Another method of avoiding any trouble from silica consists in using sulfuric and a little nitric acid as the solvent for the original substance. Silica is not soluble in hot concentrated sulfuric acid. The volume of the sulfuric should be about ten times that of the nitric acid used, and the boiling be continued until sulfuric vapors are evolved. [Pg 54]
2. Error Due to Arsenic.—Only in rare cases will arsenic be found in phosphatic fertilizing materials. In case of pyritic phosphates, the iron disulfid may carry arsenic. The solution in such a case is best accomplished in hydrochloric acid. If aqua regia be used, all nitric acid should be removed by repeated evaporation with hydrochloric. The arsenic can then be precipitated in the hot dilute hydrochloric acid solution by hydrogen sulfid.
3. Error Due to Occluded Magnesia.—The danger of contamination of the yellow precipitate with magnesium oxid has been pointed out by some authors. The re-solution of the precipitate followed by a second precipitation is the usual remedy proposed. Lorenz states that this source of error may be entirely avoided by the addition of two per cent of citric acid to the phosphomolybdate solution.[45]
4. Error Due to Volatility of Phosphoric Acid.—This source of error has been made the subject of a special study by Neubauer.[46]
From the results, a table has been constructed, the use of which is recommended for phosphoric acid determinations. The source of error in this method lies exclusively in the loss of phosphoric acid by volatilization. The magnesia-covered crucible lid offers a very good control of this error, and its use is recommended to the analyst. Of course, the presence of sulfur in the gas used for ignition is liable to disturb this check.
The following course of procedure in the determination of phosphoric acid can be recommended to avoid or correct this error:
Separate the phosphoric acid in the form of the yellow precipitate and wash this latter in the usual way. Too high a heat should not be employed, nor should the solutions be allowed to stand too long lest excess of molybdic acid separate. Dissolve the phosphomolybdate in 100 cubic centimeters of cold two and five-tenths per cent ammonia and add as many cubic centimeters of the usual magnesia mixture (fifty-five grams magnesium chlorid and seventy grams ammonium chlorid dissolved in a liter of two and five-tenths per cent ammonia) as there are centigrams of phosphorus pentoxid present. Addition should not be made faster than ten cubic centimeters per minute. Stir during the addition. [Pg 55] After the precipitation, stir briskly once more and then allow to stand at least three hours. Wash with two and five-tenths per cent ammonia till the chlorin reaction disappears, dry the filter, and introduce into a well-cleaned crucible which has been thoroughly ignited. Place the lid at an angle, carbonize the filter, and gradually raise the heat, though not higher than a medium red heat, till the pyrophosphate becomes completely white. When this happens bring the blast into action and ignite to constant weight. The weight finally accepted must not change even after half an hour’s ignition. Upon this requirement especial stress must be laid. Pure magnesium pyrophosphate does not suffer any loss even after several hours’ ignition nor does a good platinum crucible. To the weighed amount of pyrophosphate, add the correction given in the table. For example, if the weight be 250 milligrams, the correction to be added is four and two-tenths milligrams, and the correct weight is then 254.2 milligrams. Multiplication of the sum by sixty-four gives the amount of phosphorus pentoxid in the weight taken for analysis.
Correction for Phosphoric Acid Determination.
| Found, Mg₂P₂O₇ in grams. |
Lost, milligrams Mg₂P₂O₇ |
Found, Mg₂P₂O₇ in grams. |
Lost, milligrams Mg₂P₂O₇ |
|---|---|---|---|
| 0.10 | 0.6 | 0.24 | 4.0 |
| 0.12 | 0.8 | 0.25 | 4.2 |
| 0.14 | 1.2 | 0.26 | 4.6 |
| 0.15 | 1.4 | 0.27 | 5.0 |
| 0.16 | 1.6 | 0.28 | 5.5 |
| 0.17 | 2.4 | 0.29 | 6.1 |
| 0.18 | 2.6 | 0.30 | 6.8 |
| 0.19 | 3.2 | 0.31 | 7.6 |
| 0.20 | 3.5 | 0.32 | 8.6 |
| 0.21 | 3.6 | 0.33 | 9.6 |
| 0.22 | 3.8 | 0.34 | 10.6 |
When phosphoric acid is to be estimated as pyrophosphate it must always be first separated as molybdate, even when the original solution contains no bases capable of forming insoluble phosphates, as otherwise these corrections will not be applicable.
Using these corrections the estimation of phosphoric acid becomes one of the most accurate of known analytical methods.
61. The Color of the Magnesium Pyrophosphate.—After the final ignition of the magnesium pyrophosphate, whether secured by the citrate [Pg 56] or the molybdic method, a black or grayish tint is often noticed. This may be due to traces of organic matter brought down by the precipitate and especially to a lack of care in the initial ignition. Many devices have been proposed for the purpose of avoiding this coloration, although general experiments have shown that there is no appreciable increase in the weight of the precipitate when colored in this way.
When the precipitation is carried on according to the citrate method, Neubauer[47] proposes to eliminate this coloration by the use of ammonium sulfate. About seven cubic centimeters of a saturated solution of ammonium sulfate should be added to the solution before the precipitation by the magnesium mixture. With this precaution it is possible to obtain a perfectly white precipitate after five minutes of ignition. The lively glowing of the precipitate throughout the whole mass at the time of changing into pyrophosphate, is much more easily observed by this treatment than when the mass is gray or black. Even should the addition of the ammonium sulfate solution to one containing a large amount of lime produce a precipitate of crystalline calcium sulfate, it is of no importance inasmuch as the ammonium citrate immediately dissolves large quantities of the calcium salt.
In this laboratory a white pyrophosphate is easily obtained by treating the precipitate on the gooch after washing free of chlorids with a drop or two of ammonium nitrate. The ignition is commenced very gently at first and afterwards when the mass is white the blast is used.
If the ignited residue be gray it may sometimes be whitened by moistening with a drop or two of nitric acid, burning at a very low temperature, followed by the blast. There is no appreciable difference in weight between a gray and white pyrophosphate.
62. Determination of Phosphoric Acid and Nitrogen in the Same Solution by Treatment with Sulfuric Acid and Mercury.—Fertilizing materials which contain organic nitrogen and phosphoric acid, such as bones, are of such a nature that it is often difficult to obtain a fair sample of them in quantities suited to the direct determination; viz., about one gram. Thus it often becomes important to take a [Pg 57] much larger quantity of the material, to bring it into solution and to take an aliquot part thereof. It may also often happen that it is important to determine the phosphoric acid in the same sample which has been used for the determination of the nitrogen by moist combustion with sulfuric acid and mercury. In this connection, however, it is somewhat difficult to avoid the precipitation of some of the mercury with the phosphoric acid.
The mercuric sulfate which is produced by the Kjeldahl method is not precipitated in the presence of ammoniacal solution of ammonium citrate, but there may be small quantities of mercurous salts present or some finely divided metallic mercury which may contaminate mechanically the phosphate precipitate. These disturbing influences may be removed by previous treatment with sodium chlorid. If from fifty to sixty cubic centimeters of sulfuric acid have been used for the solution and oxidation and this be made up to half a liter, it will be sufficiently dilute to permit an almost quantitative separation of the mercurous chlorid produced by treatment with sodium chlorid.
Neubauer, who has proposed this method, finds that when sodium chlorid is used previous to the precipitation of the phosphoric acid, a precipitate of ordinary size contains, at most, only one milligram of mercury, while without the use of sodium chlorid as much as four milligrams may be found. The details of the method employed by Neubauer are as follows:
Ten grams of the fertilizing material are placed in a half liter flask with from fifty to sixty cubic centimeters of strong sulfuric acid, two grams of mercury, and a little paraffin to prevent foaming. The oxidation is carried on as usual in the Kjeldahl method. The liquid, after cooling, is diluted with water and one cubic centimeter of a citrate solution of sodium chlorid added, cooled, filled to the mark, filtered, and fifty cubic centimeters taken for the determination of the phosphoric acid, according to the citrate method and the same quantity for the determination of the ammonia by distillation.
63. General Principles.—It has been seen that in the molybdic method there is introduced a process at considerable cost, both of [Pg 58] reagents and time, having for its object the separation of the phosphoric acid from all the other acids and bases which may have been present in the original sample. The phosphorus is thus obtained in composition with molybdenum and ammonium in a form easily soluble in ammonia, from which it can be accurately separated by means of a soluble salt of magnesia.
The citrate method has for its object the suppression of this intermediate step and the determination of the phosphoric acid by direct precipitation in presence of iron, lime, and alumina. The principle on which it is based rests on the well-known power of an alkaline ammonium citrate to hold in solution the salts of iron, alumina, and lime, while at the same time it permits of the separation of phosphoric acid, as ammonium magnesium phosphate. In no case can the citrate method be regarded as an exact analytical process, but large experience has shown that the errors of the method are compensatory and that it affords a good and ready method for fertilizer control.
When phosphoric acid solutions which contain no iron, lime, alumina, or manganese, are precipitated in presence of ammonium citrate the results obtained vary markedly with the quantity of magnesia mixture employed. Grupe and Tollens[48] were the first to point out that a portion of the phosphoric acid might remain in solution, but that the precipitate might contain a sufficient excess of magnesia to compensate for the loss. It has been further shown by Glaser[49] that a portion of the phosphoric acid may be lost by volatilization in the citrate method. When the ignition is carried on in a crucible where the cover is coated with magnesia to intercept the volatilized acid, a considerable quantity of it can be recovered by the molybdic method.
Where too little magnesia mixture is employed, therefore, two sources of loss are to be guarded against; viz., a part of the phosphoric acid may remain in solution and another part be volatilized on ignition. The explanation of the volatilization is as follows: In the presence of ammonium citrate, magnesium chlorid may be partly converted into magnesium citrate and ammonium chlorid. There may be a time, therefore, in the precipitation with not too great excess of magnesia mixture, when proportionally there is little magnesium [Pg 59] chlorid and much ammonium chlorid present. The formation of a salt represented by the formula Mg(NH₄)₄(PO₄)₂ may take place which, upon ignition, breaks up into Mg(PO₃)₂ and finally passes into Mg₂P₂O₇ with loss of P₂O₅. This theoretical condition has but little weight, however, practically in the analysis of fertilizers, since in these cases a large quantity of lime is always present. But even in these cases traces of volatile P₂O₅ may be discovered.
Wells[50] has shown that the citrate method gives good results in certain conditions but that this accuracy is reached by a fortunate compensation of errors. The ammonium magnesium salt does not precipitate all the phosphoric acid in this process, but contains enough impurities to make up for this loss.
Johnson[51] in conjunction with Osborne has shown that the results by the citrate method practiced in accordance with the details laid down by Vögel, are too low, but that this difficulty could be overcome by using more and stronger magnesia mixture and a larger quantity of strong ammonia solution. The citrate method was found to give unsatisfactory results when iron and alumina were present in any considerable quantity. In the examination of the final ignited precipitate, which should be pure magnesium pyrophosphate, it was found to consist of only 94.98 to 97.83 per cent of that salt. The chief impurity found was calcium oxid, the percentage of which varied from 2.05 to 3.95 in six cases. There was also a considerable percentage of loss due, probably, to magnesia and pyrophosphoric acid.
The presence of large quantities of iron and alumina also impairs the accuracy of the molybdate method when the precipitation of the yellow salt takes place at too high a temperature. When the temperature of precipitation in the method is above 50° the results are likely to be too high while a great excess of nitric acid in the reagent may produce a contrary effect. In the latter case the filtrate from the yellow salt should be mixed with additional quantities of molybdate solution until no further precipitate takes place.
Many methods of conducting the citrate method have been proposed but the best of them are based on the one elaborated at the experiment station of Halle by Bühring, and which will be given in the next paragraph, followed by some other methods in use in other localities. [Pg 60]
64. Method of the Halle Agricultural Experiment Station.[52]—The citrate method, as described by Morgen, is the one employed.[53] The principle depends upon the direct precipitation of the phosphoric acid by magnesia mixture. By the addition of a solution of ammonium-citrate the precipitation of lime, iron, alumina, and other bases, is prevented. The precipitate of ammonium magnesium phosphate is converted by ignition into magnesium pyrophosphate and weighed as such. By the use of this method a part of the phosphoric acid sometimes escapes precipitation and a portion of the other bases is sometimes thrown down with the precipitate. Experience has shown that by adhering to certain precautions the weight of impurities in the precipitate may be made to correspond exactly to the weight of the phosphoric acid which escapes precipitation.
Figure. 3.
Shaking Apparatus for Superphosphates.
(1) Soluble Acid.—The soluble phosphates are first brought into solution in such a way that one liter of water contains the soluble phosphoric acid from twenty grams of the substance. Twenty grams are rubbed in a porcelain mortar with water and through a wide-necked funnel washed into a bottle-shaped flask in which a little water has been previously placed. The flasks employed are made of thick glass in order to withstand shaking. After the substance is washed, the flasks are filled to the mark and closed with rubber stoppers. They are then placed upon a shaking rack as indicated in Fig. 3, [Pg 61] which is also furnished with an apparatus for separating the fine meal from basic slag.
On a table, as shown in the figure, is fastened a movable horizontal board by means of hinges. At the left hand of this movable board is placed an open wooden box in which is a perforated shelf for the purpose of holding the flasks, so as to prevent their striking together during the shaking.
For the best results the substance to be examined should be placed in the flask in a dry state and then 800 cubic centimeters of water added and shaken by means of the machine indicated for half an hour. Afterwards the flasks are filled up to the mark, well shaken, and filtered through double folded filters into ordinary flasks of about 400 cubic centimeters capacity. Before any of the filtrate is collected, the first that runs through should be well shaken in the receiving flasks and rejected. Fifty cubic centimeters of the filtrate thus collected, corresponding to one gram of the substance, should be used for the determination.
(2) Total Acid.—For total phosphoric acid, including the insoluble portions, the material is treated as follows: Five grams of the substance are placed in a 500 cubic centimeter flask with twenty cubic centimeters of nitric acid of 1.42 specific gravity, and fifty cubic centimeters of pure concentrated sulfuric acid, and boiled briskly for half an hour. With substances which contain much organic material, a little paraffin is added to avoid frothing. Such substances also require a larger quantity of nitric acid than that above specified. The flasks are allowed to cool, water added, again allowed to cool, and filled up to the mark at 17°.5. If hydrochloric instead of sulfuric acid be used in making the above solution, when the citrate method is employed, the results are always too high because the precipitate contains lime and alumina in such quantities as to render any compensation for them inaccurate. In addition to this the sulfuric has this great advantage over the hydrochloric acid; viz., in not separating silicic acid, inasmuch as the silicic acid is insoluble in boiling sulfuric acid.
(3) Citrate-Soluble Acid.—Two grams of the sample are digested with 100 cubic centimeters of citrate solution, 1.09 specific gravity, for half an hour at 50° in a beaker. Afterwards the soluble matter is separated by filtration with the aid of a filter-pump and the residue [Pg 62] washed with a solution of one part water and one part citrate solution until all the dissolved phosphoric acid is removed from the filter. Generally three or four washings are sufficient. The residue on the filter is dried, ignited, and dissolved in a mixture of two cubic centimeters of nitric and twenty cubic centimeters of sulfuric acid, the solution made up to a volume of 200 cubic centimeters, filtered, and 100 cubic centimeters of the filtrate taken for the determination. The acid in the filtrate is nearly neutralized and fifty cubic centimeters of citrate solution are added, and afterwards twenty-five cubic centimeters of magnesia mixture and twenty cubic centimeters of twenty-four per cent ammonia. After standing for forty-eight hours, the precipitate is separated by filtration, ignited, and weighed in the usual way. The difference between the total phosphoric acid and that in the insoluble residue, after treatment with ammonium citrate, as above, gives the quantity of phosphoric acid soluble in the citrate solution. The difference between the total citrate-soluble and the water-soluble gives the quantity of the reverted phosphoric acid.
The ammonium citrate solution used for the digestion is made as follows: Two hundred and fifty grams of crystallized citric acid are dissolved in half a liter of hot water, diluted with 550 cubic centimeters of water, 276 cubic centimeters of twenty-four per cent ammonia added, and finally, exactly neutralized by adding, little by little, fifty per cent citric acid solution.
The Halle methods of separating the water and citrate-soluble acids appear to be less complete and reliable than those in use by the Official Agricultural Chemists of this country. The precipitation of basic phosphates, when large quantities of water are used at once in separating soluble acid, must tend to diminish the quantity obtained, while the lack of care in assuring the neutrality of the citrate solution might lead to varying results.
(4) Double Superphosphates.—In the case of double superphosphates, which sometimes contain large quantities of pyrophosphate, the solution is made in the usual way so that in 100 cubic centimeters there will be contained two grams of the substance. Usually ten grains are taken and the volume made up to half a liter. Twenty-five cubic centimeters of the filtrate are diluted with seventy-five cubic centimeters of water and the pyro converted to [Pg 63] orthophosphoric acid by heating with ten cubic centimeters of strong nitric acid on a sand-bath. The heating should be continued until the volume be reduced to twenty-five cubic centimeters. The strongly acid liquid is made alkaline with ammonia, and afterwards slightly acid with nitric, and the rest of the process is carried on in the usual way.
(5) Phosphoric Acid in the Residue of Superphosphate Manufacture.—In the mixture of superphosphates and gypsum, the residue of the manufacture of double superphosphates, the phosphoric acid is estimated in the following manner: Five grams of the substance are placed in a dish, rubbed up with absolute alcohol, and washed into a 250 cubic centimeter flask. The flask is filled with absolute alcohol to the mark, closed with a stopper, and with frequent shaking, allowed to stand for two hours; it is thereupon filtered as quickly as possible; fifty cubic centimeters of the filtrate corresponding to one gram of the substance, are taken for the estimation. This fifty cubic centimeters is evaporated on a sand-bath to a sirupy consistence, diluted with water, and treated, as in the case of the soluble phosphates above mentioned. In all cases as described above, after the solutions are obtained they are treated with the ammonium citrate solution and the phosphoric acid estimated as in the first instance given.
(6) Solutions Employed.—
(a) The citrate solution is made as follows: 1,500 grams of citric acid are dissolved in water, treated with five liters of twenty-four per cent ammonia, and made up to fifteen liters.
(b) The magnesia mixture is made as follows: 500 grams of magnesium chlorid, 1,050 grams of ammonium chlorid, three and five-tenths liters of twenty-four per cent ammonia, and six and five-tenths liters of distilled water are used.
In the case of the superphosphates fifty cubic centimeters of the citrate solution are employed and with the basic slags 100 cubic centimeters; and in both cases twenty-five cubic centimeters of the magnesia mixture.
(7) Details of the Manipulation.—On the addition of the citrate solution there should be no permanent troubling of the liquid but there should be a total clearing up thereof. In order to facilitate this, after the addition of the citrate solution, the flasks should be gently shaken in order to distribute the solution throughout the mass. [Pg 64] Solutions from bone-black superphosphates show sometimes, after the addition of the citrate solution, a more or less strong opalescence, but this opalescence does not influence the results. Should it happen that with superphosphates which are made from raw material containing large excesses of iron or clay, fifty cubic centimeters of the citrate solution are not sufficient to prevent the other bases from being precipitated, an additional quantity up to twenty-five cubic centimeters may be added. The addition of the magnesia mixture must follow as quickly as possible after the addition of the citrate solution to avoid a separation of crystalline calcium phosphate. On the addition of the citrate solution there is always a rise in temperature. Inasmuch as the precipitation of the phosphoric acid with magnesia must take place in the cold, the liquid must be cooled after the addition of the citrate,[54] and the cooling should take place as quickly as possible.
The above method was adopted by the chemical section of the International Agricultural Congress held at Vienna, September, 1890.[55]
Figure. 4.
Shaking Machine for Ammonium Magnesium Phosphate.
In order to hasten the precipitation of the ammonium magnesium phosphate and to prevent the fixation of the precipitate on the walls of the erlenmeyer, the flask should be shaken for half an hour. For this purpose the flasks should be closed with smooth well-fitting rubber stoppers and placed in a shaking machine. The shaking machine of the form given in Fig. 4, recommended by the Halle station, is very conveniently used for this purpose. [Pg 65]
On a vertical axis are carried two stages for holding the flasks. The flasks are prevented from striking each other by means of the partitions shown. The apparatus is conveniently driven by a small water-motor, as indicated, which imparts to the stages a partial back and forth revolution.
After shaking for half an hour, any precipitate adhering to the rubber stoppers is carefully washed off with ammonia water into the flask. The filtration can be made immediately after the shaking or after two or three days; the results are the same.
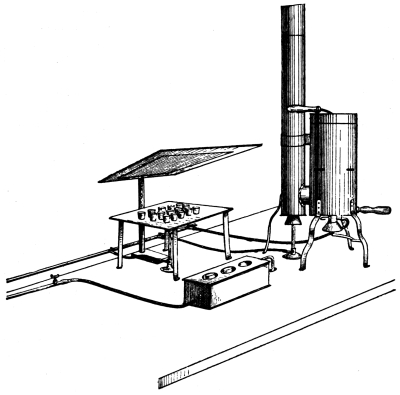
Figure. 5.
Rössler Ignition Furnace.
The filtration of the ammonium magnesium phosphate is made through perforated crucibles. The asbestos felt is prepared in the following way: The coarse fibers of asbestos are chopped up with a sharp knife on a glass plate and boiled for two hours with strong hydrochloric acid; [Pg 66] afterwards, by repeated washing with distilled water they are freed from acid and the too fine particles of asbestos which would tend to make the filter too impervious. After the last wash-water is poured off, the asbestos is suspended in water and used for making the felt on the filter. The preparation of the crucible and the filtration under pressure are accomplished in the usual way.
The ignition of the precipitate is accomplished in a Rössler ignition oven, Fig. 5. When the muffle of the furnace shows a white heat or a white-red heat it is at the proper temperature for the estimation. At higher temperatures, the asbestos felt is easily injured. Generally, an ignition of five minutes is sufficient, but with double superphosphates, ten minutes are required.
65. The Swedish Citrate Method.[56]—This method of determining phosphoric acid is founded on the fact that phosphoric acid in the presence of calcium salts, without it being necessary, previously, to convert it into phosphomolybdate, is precipitated directly by magnesia mixture from a solution, to which ammonium citrate has been added, provided first, that the solution contain a sufficient quantity of sulfuric acid, and second, that only as much citrate be added as is required to keep the calcium salts in alkaline solution.[57]
Reagents. (1) Citric Acid Solution.—Prepared by dissolving 500 grams of citric acid in water, and completing to a volume of one liter.
(2) Ten Per Cent Ammonia of 0.959 specific gravity.
(3) Magnesia Mixture, of the usual composition.
The various processes are conducted as follows:
(a) Water-Soluble Phosphoric Acid.—Add twenty cubic centimeters of citric acid solution to fifty cubic centimeters of the water-soluble solution obtained according to the Swedish molybdenum method, and then add thirty-three cubic centimeters of ammonia. When the mixture has cooled, add slowly twenty-five cubic centimeters of the magnesia mixture, and then forty-two cubic centimeters of the ammonia. Keep the solution stirred by means of a closely clipped feather which is pressed tightly against the sides of the beaker; by this process the phosphate is precipitated after half an hour in pure condition and completely, without, in the least, sticking to the wall of the beaker; filter, wash, and ignite, as usually directed. [Pg 67]
(b) Insoluble Phosphoric Add.—Moisten, in a porcelain dish, ten grams of the powdered sample with water; add fifty cubic centimeters of concentrated sulfuric acid, and heat for fifteen minutes so high that fumes of sulfuric acid will escape. When the mass has cooled, wash it into a half liter graduated flask, fill to the mark, and shake well. After filtration, the clear filtrate may, after some time, turn turbid by separation of calcium sulfate, but as the ammonium citrate, which is afterwards added, again brings the precipitate into solution, it is of no importance. Take fifty cubic centimeters of the solution, corresponding to one gram of the powdered sample, add twenty cubic centimeters of the citric acid solution, neutralize the mixture approximately, but not exactly, by ammonia; after cooling, add twenty-five cubic centimeters of magnesia mixture; stir the fluid by means of a feather, as described above, till no more precipitate is formed, and finally add thirty-three cubic centimeters of ammonia while stirring for a couple of minutes more; after half an hour the precipitate may be separated by filtration, washed, and ignited, as usually directed.
The above process is essentially the one used with basic slags. When much organic matter is present, by continuing the heating with sulfuric acid for some time, it may be destroyed.
66. Methods Adopted by the Brussels Congress, 1894.—The report of the committee on methods of analysis of phosphoric acid requires the molybdate method to be used in all cases where the quantity to be determined is very small. In other cases the citrate method may be employed.[58]
(1) Soluble Phosphoric Acid.—The soluble phosphoric acid is determined by the method adopted at Brussels in the following manner: Five grams of the sample are rubbed to a powder in a mortar, and then from fifty to sixty cubic centimeters of water added. After allowing to settle for a few minutes the liquid portion is decanted upon a filter. This operation is repeated three or four times. Finally the solid portions are washed upon the filter, and the washing with water is continued until the filtrate amounts to about three-quarters of a liter. A few drops of hydrochloric acid are added until the filtrate is perfectly clear, and the volume is then made up to one liter. Fifty [Pg 68] cubic centimeters of the solution are then treated with thirty cubic centimeters of ammonium citrate solution and one-third as much ammonia. Afterwards thirty cubic centimeters of magnesia mixture are added, drop by drop, with constant stirring.
For superphosphates containing more than eighteen per cent of phosphoric acid only one gram is taken, for ordinary superphosphates two grams, and for compound fertilizers four grams. The sample is first treated as above for soluble acid until the filtrate amounts to 200 cubic centimeters, then clarified with a drop of nitric acid, and made up to a quarter of a liter.
(2) Reverted Phosphoric Acid.—The filter containing the residue is then introduced into a quarter liter flask and treated with 100 cubic centimeters of Petermann’s alkaline ammonium citrate solution, vigorously shaken, and left at room temperature for fifteen hours. It is then digested for an hour at 40° and filtered. Fifty cubic centimeters of the filtrate are placed in a flask and, with constant shaking, thirty-five cubic centimeters of magnesia mixture added. The aqueous solution is treated in the same way. The precipitate is collected, ignited and weighed, and multiplied by 0.64 for phosphoric acid. The total acid is determined in the usual way.
67. Dutch Method for Citrate-Soluble Phosphoric Acid.[59]—The reagents necessary are:
(1) Citrate solution, prepared according to Petermann. Dissolve 165 grams of citric acid in 700 cubic centimeters of water, mix with 250 cubic centimeters of ammonia of 0.92 specific gravity, and, after cooling, bring to the volume of one liter.
(2) Magnesia mixture prepared according to Petermann. Dissolve 400 grams of crystallized magnesium chlorid, 800 grams of ammonium chlorid, and 1,600 cubic centimeters of ammonia of 0.96 specific gravity in water, and dilute to five liters.
The quantity to be taken for the analysis is five grams where the fertilizer contains less than six per cent of phosphoric acid (mixed fertilizers); two grams where it contains more than six and less than fifteen per cent (common superphosphates); and one gram where it contains more than fifteen per cent (double superphosphates). Place the weighed substance in a mortar and cover with 100 cubic centimeters of [Pg 69] citrate solution. Gently rub up, wash into a half liter flask, and heat in a water-bath for an hour to a temperature between 35° and 38°. Allow to cool, fill up to 500 cubic centimeters, and filter through a dry double filter. If it is not clear at the first filtration, pour through the filter again, repeating this till clearness is attained. Measure 100 cubic centimeters and add seventy-five cubic centimeters of magnesia mixture, allowing the latter to flow into the former very slowly, and constantly stirring during the influx. Allow to stand fifteen hours, filter, wash with ammonia of 0.96 specific gravity, dry, ignite, and weigh.
The per cent of phosphoric acid, except where otherwise indicated, is always to be given as per cent of anhydrous acid (P₂O₅).
68. Comparative Accuracy of the Citrate and Molybdate Methods.—The general use of the citrate method of determining phosphoric acid by the German chemists has led Johnson[60] to review some trials of that method in the Yale laboratory made as early as 1880. These determinations have lately been repeated in comparison with the ordinary molybdate methods with the result that in sixty-seven determinations on bone-dust, superphosphate, cotton-hull ashes, cottonseed-meal, tankage, bone-char, phosphatic guano, and phosphate rock, only three citrate results differed from those obtained by the molybdate method by more than three-tenths of one per cent. The greatest discrepancy between the two methods was 0.41 per cent, and the average difference was 0.09 per cent.
The citrate method was found to give poor results when iron and alumina were present in considerable quantity. Ignited precipitates by the citrate method were found to contain as high as four per cent of lime, and iron and alumina in small quantities when these bodies were abundant in the original substance.
In the molybdate method the rapid precipitation from solutions at 65° was found to give unsatisfactory results and it was found necessary to conduct the process at temperatures between 40° and 50°. With a relative excess of nitric or a relative deficiency of molybdic acid some phosphoric acid may easily escape precipitation. The chief [Pg 70] objection to precipitating at 65° is found in the fact that in presence of considerable iron and alumina some of these bodies may be found in the yellow precipitate, whence they pass to the final ammonium magnesium phosphate.
The citrate method, therefore, only gives safe results by compensating errors which in every class of phosphates must be empirically determined.
The molybdate method gives results too high when iron and alumina are present in considerable quantity and the yellow precipitate is obtained at temperatures above 50°. On the other hand, if there, be a great relative excess of nitric acid the results may be too low unless the filtrates from the yellow precipitate be mixed with additional molybdic solution and digested until no further precipitate is formed.
Comparative determinations made, by both methods, by the Association of German Experiment Stations have led to the conclusion that both give practically the same results when each one is conducted with the proper precautions peculiar to it.[61] In the latter part of 1892, at the general meeting of the Association, it was declared that the citrate method, after having been subjected to repeated tests, was found to be satisfactory, changing the composition of the solution so that it might have 1,100 instead of 1,000 grams of citric acid and four liters of twenty-four per cent ammonia to each ten liters. The data afforded by the citrate method, when applied to an artificial mixture of known composition, were more satisfactory than those obtained by the molybdic process.
In this laboratory the citrate method has been found to give nearly agreeing results with the old process. It is much shorter and less expensive; and is recommended most favorably for practical use, suggesting, however, that with every new kind of phosphate or phosphatic fertilizer varying notably in composition from the standard, the work should be checked at first by comparison with the molybdenum method.
69. History and Manufacture.—The basic process for the manufacture of Bessemer steel is known in Europe as the Thomas [Pg 71] or Thomas and Gilchrist process, and the slags rich in phosphate, one of the waste products of the process, are known by the same name. In this country all the phosphatic slags which have been made in the manufacture of steel have been obtained working under the patents of Reese, and, when prepared for the market, are known as odorless phosphate. The only place where these slags have been made in this country is Pottstown, Pennsylvania. In Europe they are extensively manufactured, in England, France, and Germany, and their use for agricultural purposes has increased until it is quite equal to that of superphosphates.
The quantity of basic slag manufactured in Germany in 1893 was 750,000 tons; in England 160,000; in France 115,000, making the total production of central Europe about 1,000,000, a quantity sufficient to fertilize nearly 5,000,000 acres.
70. Process of Manufacture.—The principle of the process depends upon the arrangement of the furnaces, by means of which the phosphoric acid in the pig iron is caused to combine with the lime which is used as a flux in the converters. A general outline of the process is as follows:
The pigs, which contain from two to four per cent of phosphorus, are melted and introduced into a Bessemer converter lined with dolomite powder cemented with coal-tar, into which has previously been placed a certain quantity of freshly-burned lime. For an average content of three per cent of phosphorus in the pig iron, from fifteen to twenty pounds of lime are used for each 100 pounds of pig iron. As soon as the melted pig iron has been introduced into the converter, the air-blast is started, the converter placed in an upright position, and the purification of the mass begins. The manganese in the iron is converted into oxid, the silicon into silica, the carbon into carbon dioxid and oxid, and the phosphorus into phosphoric acid.
By reason of the oxidation processes, the whole mass suffers a rise of temperature amounting in all to about 700° above the temperature of the melted iron. At this temperature the lime which has been added, melts, and, in this melted state, combines with the phosphoric acid, and the liquid mass floats upon the top of the metallic portion, which has, by this process, been converted into steel. [Pg 72]
As soon as the process, which occupies only about fifteen minutes, is completed, the fused slag is poured off into molds, allowed to cool, broken up, and ground to a fine powder. For each five tons of steel which are made in this way, about one ton of basic slag is produced.
In another process, in order to make a slag richer in phosphoric acid, a lime is employed which contains a considerable percentage of phosphate. Although the slag thus produced is richer in phosphoric acid, it is doubtful whether it is any more available for plant growth than that made in the usual way with lime free from phosphoric acid. In other words, when a basic slag is made with a lime free from phosphoric acid, nearly the whole of the phosphoric acid is combined as tetrabasic calcium phosphate. On the other hand, when the lime employed contains some of the ordinary mineral phosphate the basic slag produced becomes a mixture of this mineral phosphate with the tetracalcium salt. The mineral phosphate is probably not rendered any more available than it was before.
It is easily seen from the above outline of the process of manufacture, that basic slags can have a very widely divergent composition. When made from pig iron poor in phosphorus, the slag will have a large excess of uncombined lime and consequently the content of phosphoric acid will be low. When made from pigs rich in phosphorus there may be a comparative deficiency of lime, and in this case the content of phosphoric acid would be unusually high.
It is found also that the content of iron in the slag varies widely. In general, the greater the content of iron the harder the slag and the more difficult to grind. If the pig iron contain sulfur, as is often the case, this sulfur is found also in the slag in combination with the lime, either as a sulfid or sulfate.
No certain formula can therefore be assigned to basic slags and the availability of each one must be judged by its chemical composition.
71. Composition of Basic Slag.—The slags produced by the method above outlined may be amorphous or crystalline. When large masses are slowly cooled the interior often discloses a crystalline composition. [Pg 73] In some samples analyzed in this laboratory the crystals were found to be of two forms; viz., acicular and tabular.[62] They had the following composition:
Calculated Per Cents as
| CaO. | Fe₂O₃. | Al₂O₃. | MgO. | V₂O₂ . | P₂O₅. | SiO₂. | |
|---|---|---|---|---|---|---|---|
| Acicular crystals | 42.69 | 20.98 | 3.71 | 0.49 | 0.18 | 27.06 | 4.96 |
| Tabular crystals | 53.61 | 9.64 | 0.91 | 0.08 | —— | 33.92 | 1.75 |
These data show that the two sets of crystals belong to two distinct mineral forms. The presence of vanadium in one of the samples is worthy of remark, and leads to the suggestion that in the slags made of phosphoriferous pigs may be found any of the rare metals which may exist in the ores from which the pigs were made. The amorphous portions may have a widely varying composition and consequent content of phosphoric acid. In all good slags, however, whether in crystalline form or as amorphous powder, the lime and phosphoric acid will be found combined as tetracalcium phosphate (Ca₄P₂O₉).
72. Molecular Structure of Tetracalcium Phosphate.—Several theories have been advanced in respect of the atomic arrangement of the elements contained in a molecule of tetracalcium phosphate. It must be confessed that so little is known concerning the reactions of this body as to make theories of its constitution largely visionary. But the existence in definite crystalline form of this salt shows that it is not merely an intimate mechanical mixture, but a true molecular form. As a type of the supposed arrangement of its particles the graphic formula proposed by Kormann may be consulted; viz.,
The crystals of this salt, as may be seen by inspection of the analytical data, contain other bodies than calcium, oxygen, and phosphorus. It would be of interest to push the investigation of their constitution further and see if crystals of pure tetracalcium phosphate could be obtained, and under what conditions they would be contaminated [Pg 74] by other metallic oxids. Usually, by the color of the crystals, it will be easy to determine something of the nature, if not the extent of the contamination.
73. Solubility of Phosphatic Slags.—The high agricultural value of basic slags led to an early study of their solubility in ammonium citrate, citric acid, and other organic solutions. Even finely ground mineral phosphates and bones are soluble to some extent in ammonium citrate, as was pointed out as long ago as 1882.[63] The most common solvents used for basic slags are ammonium citrate and citric acid. The ammonium citrate should be the same as that used for the determination of reverted phosphoric acid and the citric acid solution commonly used contains five grams in a hundred cubic centimeters. The slags of different origin and even of different age vary greatly in respect of the quantity of soluble matter they contain. It is believed, however, that a very fair idea of the agricultural value of a slag may be obtained by determining its degree of solubility in one of the menstrua named.
74. Separation by Sifting.—The relative availability of a slag, as in the case of a mineral phosphate, is determined by the percentage of fine material it contains. Sieves of varying apertures are used to determine this percentage. A one-half millimeter or a one-quarter millimeter circular aperture is best, and the percentage of the total material passing through is determined. A method used in Germany consists in sifting the slag in a sieve twenty centimeters in diameter the meshes of which are from 0.14 to 0.17 millimeter square and which measure diagonally from 0.22 to 0.24 millimeter.
75. Solution of Phosphatic Slags.—Sulfuric acid has been found to be an excellent solvent for basic slags preparatory to the determination of phosphoric acid. There is, however, no unanimity of opinion concerning the best method or means of solution. Aqua regia and nitric acid are objected to because they may convert any phosphorus in combination with the iron into phosphoric acid and thus increase the quantity present.[64] But iron phosphid is seldom or ever found in slags and therefore this objection is not always tenable. Sulfuric acid has also been deemed objectionable because the gypsum separated is likely to carry with it some of the other substances to be determined. [Pg 75]
Hydrochloric acid is also excluded by some from the list of solvents because it dissolves so many of the foreign elements in the slag and thus tends to complicate the subsequent determination, especially of magnesia. Further than this a hydrochloric acid solution is not suited to the use of the citrate method now so commonly employed. When hydrochloric acid is used, moreover, the dissolved silica must be removed and thus the time required for making a phosphoric acid determination is much increased.
If the sample be sufficiently fine the occlusion of undissolved phosphate particles by the gypsum formed when sulfuric acid is used is not to be feared and the disturbance of volume by the gypsum is pretty nearly constant and can be allowed for. When five grams of slag are used the mean volume of gypsum in the solution is about two cubic centimeters.
76. Estimation of Total Acid.—In the determination of total phosphoric acid in a slag, twenty-five cubic centimeters of the strongest sulfuric acid are placed in an erlenmeyer having a wide neck, and with careful shaking five grams of the fine slag meal gradually added. The flask is heated over a naked flame until solution is complete. When the mass is cold it is washed into a quarter liter flask; again allowed to cool, filled with water to the mark, and two cubic centimeters of water corresponding to the volume of gypsum undissolved, are added, well mixed, and filtered. In fifty cubic centimeters of the filtrate the phosphoric acid is determined by either the molybdic or citrate methods already described.
77. Alternate Method.—The following method may also be used: Ten grams of the substance are heated with fifty cubic centimeters of concentrated sulfuric acid until white vapors have been evolved for some time. The operation lasts for about fifteen minutes and can be carried on in a half liter flask or in a porcelain dish. Without regarding the undissolved material the volume of the liquid is now made up to half a liter and filtered. The filtered liquid becomes turbid after some time through the separation of calcium sulfate, but this turbidity should not be regarded. To fifty cubic centimeters of the solution, corresponding to one gram of substance, twenty cubic centimeters of citric acid solution (500 grams citric acid to the [Pg 76] liter) are added, and it is afterwards nearly neutralized by the addition of ten per cent ammonia and the liquid, which is warmed by this operation, cooled. There are now added twenty-five cubic centimeters of the ordinary magnesium chlorid mixture and the solution stirred until turbidity is produced, one-third of its volume of ten per cent ammonia added, and again stirred for about a minute.
Instead of the addition of the citric acid and ammonia the ammonium citrate prepared as follows, may be added: 1,500 grams of citric acid are dissolved with water, made up to three liters and five liters of twenty-four per cent ammonia and seven liters of water added. The rest of the operation is carried on in the usual manner.
78. Halle Method for Basic Slag.—The total phosphoric acid is estimated at the Halle Station by the following process:[65]
Ten grams of the substance are moistened in a porcelain dish with a few drops of water and about five cubic centimeters of a one to one solution of sulfuric acid added, and after the mass has hardened, which takes place very soon, fifty cubic centimeters of concentrated sulfuric acid are added and stirred with a glass rod until it is evenly distributed throughout the whole mass. In stirring this mixture the greatest care must be taken, otherwise the substance would remain attached to the sides of the dish, which during later heating would cause loss through spurting. The complete solution now takes place after a few hours’ heating on a sand-bath. During the cooling the jelly-like mass must be stirred with a glass rod, and after it is cool, by means of a washing-bottle, gently along the sides of the dish, water is added, and when the mixture becomes hot it is again cooled and washed into a half liter flask, which is made up to the mark at a temperature of 17°.5 and filtered. When the acid filtrate stands for some time there is often a separation of gypsum which, however, does not in any way influence the subsequent analysis, which is made in the usual manner.
Fifty cubic centimeters of the filtrate, representing one gram of the original substance, are placed in an erlenmeyer. In the case of double superphosphates which often contain large quantities of pyrophosphates, twenty-five cubic centimeters of the filtrate just obtained, equivalent to five grams of the substance, are diluted with seventy-five cubic [Pg 77] centimeters of water, ten cubic centimeters of nitric acid of 1.42 specific gravity added, and heated on a sand-bath to convert the pyro into orthophosphates. The heating should be continued until the liquid is reduced to its original volume of twenty-five cubic centimeters. The strongly acid liquid is saturated with ammonia and with the addition of a drop of rosolic acid as an indicator, again acidified with nitric acid, and treated as with superphosphates.
79. Dutch Method for Basic Slag.—Heat ten grams of the sample with fifty cubic centimeters of sulfuric acid (1.84 specific gravity) till white vapors are evolved, shaking or stirring constantly. After cooling make the fluid up to 500 cubic centimeters with water, taking no account of the undissolved substance. Filter, and to fifty cubic centimeters of the filtrate add 100 cubic centimeters of the ammoniacal citrate solution, and after cooling, twenty-five cubic centimeters of magnesia mixture. Stir or shake for a sufficient time. After the lapse of two hours the precipitate is to be separated by filtration and treated in the usual manner.
80. Estimation of Citrate-Soluble Phosphoric Acid in Basic Slag.—Experience has shown that the manurial value of basic slags does not depend alone on their content of phosphoric acid. Slags may contain tri- as well as tetracalcium phosphate, and even this latter salt may exist in states of differing availability. In determining the availability of basic slag for manurial purposes its solubility in ammonium citrate is considered the best standard. But this solubility will evidently be influenced by the basicity of the sample, or in other words, by the quantity of lime present. A slag rich in calcium oxid would deport itself differently with a given ammonium citrate solution from one in which the lime had been chiefly converted into carbonate. If possible, therefore, all samples should be reduced to the same state of basicity before the action of any given solvent is determined.
Wagner proposes to neutralize the basicity of a slag in the following manner:[66] Five grams of the slag are placed in a half liter flask which is then filled up to the mark with a one per cent solution of citric acid and shaken for half an hour. After filtering, fifty cubic centimeters [Pg 78] are titrated with a standard soda solution using phenolphthalein as indicator. This gives the quantity of citric acid necessary to neutralize the slag. To a second portion of five grams of the sample in a half liter flask are added 200 cubic centimeters of water and enough five per cent citric acid solution to neutralize the lime and then 200 cubic centimeters of acid ammonium citrate made as indicated below. After filling to the mark with water it is shaken for half an hour and filtered. To fifty cubic centimeters of the filtrate are added 100 cubic centimeters of molybdic solution and heated to 80°. After cooling, the precipitate is filtered and the phosphoric acid estimated in the usual way.
The acid ammonium citrate solution used is made as follows: Dissolve 160 grams of citric acid with enough ammonia to represent about twenty-eight grams of nitrogen and make up with water to one liter. The exact method is given in 82.
The molybdic solution is made by dissolving 125 grams of molybdic acid in a slight excess of two and a half per cent of ammonia, adding 400 grams of ammonium nitrate, diluting to one liter and pouring the solution into one liter of nitric acid having a specific gravity of 1.19. After allowing to stand at room temperature for one day the mixture is filtered and is then ready for use.
81. Wagner’s Shaking Apparatus.—The latest directions given by Wagner for determining the phosphoric acid in slags and raw phosphates soluble in citrate solutions, are the following:[67] Five grams of the material as it is sent into commerce without grinding or sifting, are placed in a half liter flask and covered with nearly a quarter liter of water, and then 200 cubic centimeters of citrate solution added, prepared as described below. The flask is filled to the mark with water. The flasks, which are of the shape shown in the figure, are closed with rubber stoppers, and without delay placed for half an hour in a rotating apparatus, (Fig. 6) which is turned on its axis from thirty to forty times a minute. If a shaking apparatus be used instead of the one mentioned, 200 cubic centimeters of the citrate solution should be placed in a half liter flask, filled to the mark with water, and the contents poured into a liter flask containing the phosphate. [Pg 79] This flask should be placed in a nearly horizontal position in the apparatus and the agitation be continued for half an hour. On removal from the apparatus the mixture is filtered and fifty cubic centimeters thereof treated with double the quantity of molybdic solution at 80° and the precipitate separated after cooling. The precipitate is carefully washed with one per cent nitric acid mixture, after which the filter is broken and the precipitate washed into a beaker with two per cent ammonia and the filter washed therewith until about 100 cubic centimeters have been used. If the solution is turbid from the presence of silicic acid it should be precipitated a second time by addition of molybdic solution until the acid reaction is restored. The ammoniacal solution of the yellow precipitate is treated, drop by drop, with constant stirring, with fifteen cubic centimeters of magnesia mixture, and set aside for two hours. The precipitate is collected, washed, ignited, and weighed in the usual manner. The direct precipitation of the phosphoric acid by the magnesia solution in presence of citrate is not advisable because of the almost general presence of silicic acid which would cause the results to be too high.

Figure. 6.
Wagner’s Digestion Apparatus for Slags.
The chief objection to this method of Wagner lies in the failure to control the temperature at which the digestion with citrate solution is made. Huston has shown, as will be described further on, that the temperature exercises a great influence in digestion with citrate. [Pg 80] Since the laboratory temperature, especially in this country, may vary between 10° and 35°, it is evident that on the same sample the Wagner method would give very discordant results at different seasons of the year.
82. Solutions Employed in the Wagner Method.—1. Ammonium Citrate.—In one liter there should be exactly 150 grams of citric acid and 27.93 grams of ammonia, equivalent to twenty-three grams of nitrogen. The following example illustrates the preparation of ten liters of the solution: In two liters of water and three and a half liters of eight per cent ammonia, 1,500 grams of citric acid are dissolved and the cooled solution made up exactly to eight liters. Dilute twenty-five cubic centimeters of this solution to 250 cubic centimeters and treat twenty-five cubic centimeters of this with three grams of calcined magnesia and distill into forty cubic centimeters of half normal sulfuric acid. Suppose the ammonia nitrogen found correspond to twenty cubic centimeters of fourth normal soda-lye. Then in the eight liters are contained
of ammonia nitrogen. Then in order to secure in the ten liters the proper quantity of ammonia there must be added two liters of water containing 230 - 224 = six grams of nitrogen or seven and three-tenths grams ammonia; viz., ninety-four cubic centimeters of 0.967 specific gravity.
2. Molybdate Solution.—Dissolve 125 grams of molybdic acid in dilute two and five-tenths per cent ammonia, avoiding a large excess of the solvent. Add 400 grams of ammonium nitrate, dilute with water to one liter and pour the solution into one liter of nitric acid of 1.19 specific gravity. Allow the preparation to stand for twenty-four hours at 35° and filter.
3. Magnesia Mixture.—Dissolve 110 grams of pure crystallized magnesium chlorid and 140 grams of ammonium chlorid in 700 cubic centimeters of eight per cent ammonia and 130 cubic centimeters of water. Allow to stand several days and filter.
83. Estimation of Lime.—When the lime is to be determined in basic slags some difficulty may be experienced by reason of danger of contamination of the oxalate precipitate with iron and especially manganese, which is often present in slags.
Holleman[68] proposes to estimate the lime in basic slag by a modification of the methods of Classen and Jones. The manipulation is as follows: Fifty [Pg 81] cubic centimeters of the solution of slag, equivalent to one gram of substance, are evaporated to a small volume, twenty cubic centimeters of neutral ammonium oxalate solution (one to three) added to the residue and heated on a water-bath with frequent stirring, until the precipitate is pure white and free from lumps. The time required is usually about ten minutes. The precipitate is collected on a filter and washed with hot water until the filtrate contains no oxalic acid. The precipitated calcium oxalate must be snow-white. The filter is broken and the calcium oxalate washed through, first with water and finally with warm, dilute hydrochloric acid (one to one). The calcium oxalate is dissolved by adding fifteen cubic centimeters of concentrated hydrochloric acid, the solution evaporated to a volume of about twenty-five cubic centimeters and ten cubic centimeters of dilute sulfuric acid (one to five), and 150 cubic centimeters of ninety-six per cent alcohol added. After standing three hours or more the precipitate is separated by filtration and washed with ninety-six per cent alcohol until the washings show no acid reaction with methyl orange. The calcium sulfate precipitated is dried to constant weight. This method gives a pure precipitate of calcium sulfate, containing only traces of manganese.
84. Estimation of Caustic Lime.—The lime mechanically present in basic slags is likely to be found as oxid or hydroxid, especially when the sample is of recent manufacture. In the form of oxid the lime may be determined by solution in sugar. In this process one gram of the fine slag meal is shaken for some time with a solution of sugar, as suggested by Stone and Scheuch.[69] The dissolved lime is separated as oxalate by treatment of the solution with the ammonium salt. The calcium oxalate may be determined by ignition in the usual way or volumetrically by solution in sulfuric acid and titration of the free oxalic acid with potassium permanganate solutions. The standard solution of permanganate should be of such a strength as to have one cubic centimeter equivalent to about 0.01 gram of iron. The iron value of the permanganate used multiplied by 0.5 will give the quantity of calcium oxid found. [Pg 82]
85. Detection of Adulteration of Phosphatic Slags.—The high agricultural value of phosphatic slags has led to their adulteration and even to the substitution of other bodies. Several patents have also been granted for the manufacture of artificial slags of a value said to be an approximation to that of the by-products of the basic pig iron process.
(1) Method of Blum.—One of the earliest methods of examining basic slag for adulterations is the method of Blum.[70] This method rests upon the principle of the determination of the carbon dioxid in the sample. The basic phosphatic slag is supposed to contain no carbon dioxid. This is true only in case it is freshly prepared. The tetrabasic phosphate, after being kept for some time, gradually absorbs carbon dioxid from the air. As high as nineteen per cent of carbon dioxid have been found in slags which have been kept for a long while. When the slag has absorbed so much of carbon dioxid and water from the air as to be no longer profitable for market, it can be restored to its original condition by ignition.
(2) Method of Richter-Forster.—One of the common adulterants of tetrabasic phosphate is aluminum phosphate. The method of detecting this when mixed with the slag is described by Richter-Forster.[71] The method depends on the fact that soda-lye dissolves the aluminum phosphate, although it does not dissolve any calcium phosphoric acid from the slag. Two grams of the sample to be tested are treated with ten cubic centimeters of soda-lye of from 7° to 8° C. in a small vessel with frequent shaking for a few hours at room temperature. After filtration the filtrate is made acid with hydrochloric and afterwards slightly alkaline with ammonia. With pure basic slag there is a small trace of precipitate produced, but this is due to a little silica which can be dissolved in a slight excess of acetic acid. If, however, the basic slag contain aluminum phosphate, a dense jelly-like precipitate of aluminum phosphate is produced.
(3) Method of Jensch.—Edmund Jensch[72] determines the tetrabasic phosphate in slags by solution in organic acids, and prefers citric acid for this purpose. This method was also recommended by Blum[73].
It is well known that the tetrabasic phosphate in slags is completely soluble in citric acid while the tribasic phosphate is only slightly, if [Pg 83] at all, attacked. The neutral ammonium salts of organic acids do not at first attack the tribasic phosphate at all, and they do not completely dissolve the tetrabasic phosphate. The solution used by Jensch is made as follows: Fifty grams of crystallized citric acid are dissolved in one liter of water. A weaker acid dissolves the tetrabasic phosphate too slowly and a stronger one attacks the tribasic phosphate present.
Schucht recommends the following method of procedure:[74] One gram of the slag, finely ground, is treated in a beaker glass with about 150 cubic centimeters of Jensen’s citric acid solution and warmed for twelve hours in an air-bath at from 50° to 70° with frequent shaking. Afterwards it is diluted with 100 cubic centimeters of water, boiled for one minute and filtered. The filter is washed thoroughly with hot water and the phosphoric acid is estimated in the filtrate in the usual way. With artificial mixtures of basic slags and other phosphates the quantity of basic slag can be determined by the above method.
(4) Method of Wrampelmeyer.—According to Wrampelmeyer the most convenient method for discovering the adulteration of basic slag is the use of the microscope.[75] All finely ground natural phosphates are light colored and with a strong magnification appear as rounded masses. In basic slags the particles are mostly black but there are often found red-colored fragments having sharp angles which refract their light in a peculiar way so that, with a very little experience, they can be recognized as being distinctive marks of pure basic slag.
In artificial mixtures of these two phosphates, which we have made in our laboratory, we have been able to detect with certainty as little as one per cent of added mineral phosphate.
One form of adulterating natural mineral phosphates has been mixing them with finely pulverized charcoal or soot to give them the black appearance characteristic of the basic slags. This form of adulteration is at once disclosed by simple ignition or by microscopic examination.
(5) Loss on Ignition.—If all doubts cannot be removed by the use of the microscope, the loss on ignition should be estimated. Natural phosphates all give a high loss on ignition, ranging from eight to twenty-four per cent, while a basic slag gives only a very slight [Pg 84] loss on ignition, especially when fresh. A basic slag which has stood for a long while and absorbed carbon dioxid and moisture, may give a loss on ignition approximating, in a maximum case, the minimum loss on ignition from a natural phosphate.
In experiments made in this laboratory in testing for loss on ignition, we have uniformly found that natural mineral phosphates will lose from nearly one to two and one-half times as much on ignition as a basic slag which has been kept for two years. A basic slag in the laboratory more than two years old gave, as loss on ignition, 4.12 per cent. Several samples of finely ground Florida phosphates gave the following percentages of loss on ignition, as compared with a sample of slag.
Odorless phosphate 4.12.
Florida phosphates 8.06, 6.90, 9.58, 6.40, 10.38, and 10.67 respectively.
There are some mineral phosphates, however, which are ignited before being sent to the market. We have one such sample in our laboratory from Florida which gave, on ignition, a loss of only one and four-tenths per cent. In this case it is seen that the application of the process of ignition would not discriminate between a basic slag and a mineral phosphate.
It may often be of interest to know what part of the loss, on ignition, is due to water in form of moisture. In such cases the sample should first be dried to constant weight in a steam-bath and then ignited. In the following data are found the results obtained here with samples treated as above indicated and also ignited directly. Number one is a basic slag two years old and the others Florida phosphates.
| Heated to 100° C. then ignited. | Ignited directly. | ||||
|---|---|---|---|---|---|
| Loss at 100° C. |
Loss on ignition. |
Total loss. |
Loss on ignition. |
||
| No. 1 | (Slag) | 2.57 | 1.77 | 4.34 | 4.12 |
| No. 2 | (Rock) | 2.61 | 5.19 | 7.80 | 8.06 |
| No. 3 | “ | 1.09 | 5.77 | 6.86 | 6.90 |
| No. 4 | “ | 0.42 | 9.20 | 9.62 | 9.58 |
| No. 5 | “ | 1.81 | 4.83 | 6.64 | 6.40 |
| No. 6 | “ | 4.36 | 6.52 | 10.88 | 10.83 |
| No. 7 | “ | 3.31 | 7.01 | 10.32 | 10.67 |
(6) Presence of Sulfids.—Another point noticed in this laboratory is that the basic slags uniformly contain sulfids which are decomposed upon the addition of an acid with an evolution of hydrogen sulfid. [Pg 85]
(7) Presence of Fluorin.—In applying the test for fluorin, it has been uniformly found here that the mineral phosphates respond to the fluorin test while the basic slags, on the contrary, respond to the hydrogen sulfid test. This test, however, was applied only to the few samples we have had and may not be a uniform property.
The absence of fluorin might not prove the absence of adulteration, but its presence would, I believe, certainly prove the fact of the adulteration in that particular sample.
The fluorin test is applied by Böttcher in the following manner:[76] From ten to fifteen grams of the slag are placed in a beaker ten centimeters high and from five to six centimeters in diameter, with fifteen cubic centimeters of concentrated sulfuric acid, stirred with a glass rod, and covered with a watch-glass on the under side of which a drop of water hangs. If there be formed upon the drop of water a white murky rim, it is proof that a mineral phosphate containing fluorin has been added. After from five to ten minutes you can notice on the clean watch-glass the etching produced by the hydrofluoric acid. According to Böttcher an adulteration of ten per cent of raw phosphate in slag can be detected by this method.
(8) Solubility in Water.—Solubility in water is also a good indication, natural phosphates being totally insoluble in water, while a considerable quantity of the basic slag will be dissolved in water on account of the calcium oxid or hydroxid which it contains. If the loss on ignition is low, and the volume-weight and water-solubility high, the analyst may be certain that the sample is a pure slag.
In comparative tests made in our laboratory with a sample of basic slag and seven samples of Florida phosphate, the percentages of material dissolved by water and by a five per cent solution of citric acid were found to be as follows:
| Water-soluble. Per cent. |
Sol. in five per cent citric acid. Per cent. |
||
|---|---|---|---|
| Odorless | phosphate | 0.97 | 16.10 |
| Florida | phosphate | 0.01 | 4.15 |
| “ | “ | 0.09 | 4.66 |
| “ | “ | 0.02 | 3.43 |
| “ | “ | 0.08 | 3.61 |
| “ | “ | 0.02 | 3.79 |
| “ | “ | 0.05 | 4.46 |
| “ | “ | 0.02 | 4.24 |
[Pg 86] From the above data it is seen that the solvent action of water especially would be of value inasmuch as it dissolves only a mere trace of the mineral phosphates, approximating one per cent of the amount dissolved from basic slag. In the case of the citric acid it is found that the amount of materials soluble in this solvent for basic slag is fully four times as great as for the mineral phosphates. Both of these processes, therefore, have considerable value for discriminating between the pure and adulterated article of basic slag.
(9) Specific Gravity.—The estimation of the volume specific gravity is also a good indication for judging of the purity of the slag. This is best done by weighing directly a given volume. Basic slag will have a volume-weight of about one and nine-tenths, while natural phosphates will have about one and six-tenths.
(10) Conclusions.—From the above résumé of the standard methods which are in use for determining the adulteration of basic slag, it is seen that there are many cases in which grave doubt might exist even after the careful application of all the methods mentioned. If we had only to consider the adulteration of basic slag with certain of the mineral phosphates, that is, tricalcium phosphate, the problem would be an easy one, but when we add to this the fact that iron and aluminum phosphates are employed in the adulteration, and that artificial slags may be so used, the question becomes more involved.
In doubtful cases one after another of the methods should be applied until there is no doubt whatever of the judgment which should be rendered.
86. Classification of Methods.—The time required for a gravimetric determination of phosphoric acid has led analysts to try the speedier if less accurate processes, depending on the use of volumetric methods. The chief difficulty with these methods has been in securing some sharp method of distinguishing the end reaction. In most cases it has been found necessary to remove a portion of the titrated solution and prepare it for final testing by subsidence or filtration. [Pg 87] As is well known, this method of determining the end reaction is less accurate and more time-consuming than those processes depending on a change of color in the whole mass. All the volumetric processes now in general use may be divided into two classes; viz., (1) the direct precipitation of phosphoric acid and the determination of the end reaction by any appropriate means, and (2) the previous separation of the phosphoric acid, usually by means of a citro-magnesium or molybdenum mixture, and in the latter case the subsequent titration of the yellow ammonium phosphomolybdate either directly or after reduction to a lower form of oxidation. In respect of extent of application by far the most important volumetric method is the one depending on titration by a uranium salt after previous separation by ammoniacal magnesium citrate. A promising method after previous separation by molybdenum is the one proposed by Pemberton, but it has not yet come into general use. For small quantities of phosphoric acid or of phosphorus, such as are found in steels and irons, the method of Emmerton, either as originally proposed or as modified by Dudley and Noyes, is in frequent use. Where volumetric methods are applied to products separated by molybdic solution, the essential feature of the analytical work is to secure a yellow precipitate of constant composition. If this could be uniformly done such methods would rival the gravimetric processes in accuracy. Hence it is highly important in these methods that the yellow precipitate should be secured as far as possible, under constant conditions of strength of solution, duration of time, and manner of precipitation. In these cases, and in such only, can the quicker volumetric methods be depended on for accurate results.
The direct volumetric precipitation of the phosphoric acid by a uranium salt or otherwise is practiced only when the acid is combined with the alkalies and when iron and alumina are absent and only small quantities of lime present. This method has therefore but little practical value for agricultural purposes. In all volumetric analyses the accuracy of the burettes, pipettes, and other graduated vessels should be proved by careful calibration. Many of the disagreements in laboratories where the analytical work is conducted equally well can be due to no other [Pg 88] cause than the inaccuracy of the graduated vessels which are found in commerce. Burettes should not only be calibrated for the whole volume but for at least every five cubic centimeters of the graduation.
87. The Uranium Method.—Since the phosphoric acid of practical use for agricultural purposes is nearly always combined with lime, alumina, and iron, its volumetric estimation by means of a standard solution of a uranium salt is to be preceded by a preliminary separation by means of an ammoniacal magnesium citrate solution. The principle of the method was almost simultaneously published by Sutton,[77] Neubauer,[78] and Pincus.[79] The phosphoric acid may also be separated by means of molybdic solution or by tin or bismuth.[80] In practice, however, it has been found that when the uranium method is to be used the magnesium citrate separation is the most convenient. Since this is the method practiced almost universally in France, the method there used will be given in detail. It is based essentially on the process described by Joulie.[81]
88. Preparation of Sample.—(1) Incineration.—Since the organic matters present in a phosphatic fertilizer often interfere with the employment of uranium as a reagent, it is necessary to incinerate the sample taken for analysis.[82]
(2) Solution of the Material.—All phosphates, with the exception of certain aluminum phosphates, amblygonite for example, are easily dissolved in nitric and hydrochloric acids more or less dilute, especially on ebullition. The best solvent, however, for calcium phosphates for the uranium method is incontestably hydrochloric acid which also very easily dissolves the iron and aluminum phosphates, which are often found present with calcium phosphates.
(3) Nitric Acid.—In many laboratories nitric acid is preferred in order to avoid, in part, the solution of ferric oxid which interferes with the determination of phosphoric acid in certain processes. Since it does not act in this way for the citro-magnesium uranium method, it is preferable to employ hydrochloric acid, especially because it dissolves the iron completely and permits thus [Pg 89] the operator to judge of the success of the solvent action by the completely white color of the residue.
(4) Pyritic Phosphates.—Certain phosphates contain pyrites which hydrochloric acid does not dissolve, and there is left consequently, a residue more or less colored. In this case it is necessary to add some nitric acid and to prolong the boiling until the pyrite has disappeared, since it might retain a small quantity of phosphoric acid in the state of iron phosphate.
(5) Sulfuric Acid.—Some chemists decompose the phosphates by means of dilute sulfuric acid. This method, which is certainly able to give good results for certain products and for certain processes, presents numerous inconveniences which tend to render its use objectionable for volumetric purposes. The calcium sulfate which is formed, requires prolonged washings which lead to chances of fatal error.
If an aluminum phosphate be under examination, containing only very little or no lime, sulfuric acid is to be preferred to hydrochloric and nitric acids, since it attacks amblygonite, which, as has been before stated, resists the action of the other two acids. But these are cases which are met with very rarely, and which can always be treated by the general method by previously fusing the material with a mixture of sodium and potassium carbonate.
In the great majority of cases the decomposition by hydrochloric acid is very easily accomplished by simply boiling in a glass vessel, and without effecting the separation of the silica. This operation is only necessary after the substance has been fused with alkaline carbonates, or, in case of substances which contain decomposable silicates giving gelatinous silica with hydrochloric acid.
There are two methods [see (6) and (7)] of securing a solution of the sample taken which varies from one to five, and even ten grams, according to the apparent homogeneity of the material to be analyzed.
(6) Solution by Filtration and Washing.—The ordinary method can be employed consisting in decomposing the substance by an acid, filtering, and washing the residue upon the filter, and combining all the wash-waters to make a determinate volume. Afterwards an aliquot [Pg 90] fraction of the whole is taken for the precipitation. This method is long, and presents some chances of error, when the insoluble residue is voluminous and contains silica which obstructs the pores of the paper and renders the filtration difficult.
(7) Volumetric Solution.—It is advisable to substitute volumetric solution for solution by filtration and washing, which is accomplished by decomposing the substances in a graduated flask, the volume being afterwards made up to the mark with distilled water after cooling. The solution is then filtered without washing, and by means of a pipette an aliquot part of the original volume is taken for precipitation. Thus all retardations in the process are avoided, and likewise the chances of error from washing on the filter. It is true that this method may lead to a certain error due to the volume of the insoluble matter which is left undecomposed, but since this insoluble matter is usually small in quantity, and since it is always possible to diminish the error therefrom by increasing the volume of the solution, this cause of error is much less to be feared than those due to the difficulties which may occur in the other method. Let us suppose, in order to illustrate the above, that we are dealing with a phosphate containing fifty per cent of insoluble sand which may be considered as an extreme limit. In working on four grams of the material in a flask of 100 cubic centimeters capacity, there will be an insoluble residue of two grams occupying a volume of about one cubic centimeter, the density of the sand being generally nearly two. The one hundred cubic centimeter flask will then contain only ninety-nine cubic centimeters of the real solution, and the error at the most would be 0.01. This error could be reduced to one-half by dissolving only two grams of the material in place of four, or by making the volume up to 200 instead of 100 cubic centimeters.
In general it may be said that the errors which do not exceed 0.01 of the total matter under treatment, are negligible for all industrial products. The method of volumetric solution does not present any further inconvenience. It deserves to be and has been generally adopted by reason of its rapidity in all the laboratories where many analyses are to be made. In the volumetric method great care should be taken not [Pg 91] to make up to the volume until after the cooling to room temperature, which may be speedily secured by immersing the flask in cold water. Care should also be exercised in taking the sample for analysis by means of the pipette immediately after filtration, and filtration should take place as soon as the volume is made up to the standard. By operating in this way the possible variations from changes of volume due to changes of temperature are avoided.
(8) Examination for Arsenic Acid.—When the sample examined contains pyrites, arsenic is often present. When the decomposition has been effected by means of nitric acid, arsenic acid may be produced. This deports itself in all circumstances like phosphoric acid, and if it is present in the matter under examination it will be found united with the phosphoric acid and determined therewith afterwards. It is easy to avoid this cause of error by passing first a current of sulfurous acid through the solution, carrying it to the boiling-point in order to drive out the excess of sulfurous acid, and afterwards precipitating the arsenic by a current of hydrogen sulfid. After filtration, the rest of the operation can be carried on as already described.
89. Precipitation of the Phosphate by Magnesium Citrate.—By means of an accurate pipette a quantity of the solution representing from 0.125 to 0.250 gram or more is taken, according to the presumed richness of the product to be examined. In order that the following operations may go on well, it is necessary that the quantity of phosphoric acid contained in the sample should be about fifty milligrams. The sample being measured is run into a beaker, and there are added, first, ten cubic centimeters of magnesium citrate solution, and second, a large excess of ammonia. If the quantity of the magnesium citrate solution be sufficient, the mixture should at first remain perfectly limpid and only become turbid at the end of some moments and especially after the mixture is stirred.
If there should be an immediate turbidity produced it is proof that the quantity of magnesium citrate solution employed has been insufficient, and it is necessary to begin again by doubling its amount. Good results cannot be obtained by adding a second portion of the magnesium citrate [Pg 92] solution to the original, since the iron and aluminum phosphates which are once formed are redissolved with difficulty. Many chemists at the present time abstain from using the magnesium citrate solution and replace it by a solution of citric acid and one of magnesium sulfate, which they pour successively into the sample under examination. This is a cause of grave errors which it is necessary to point out. Joulie has indeed recognized the fact that the precipitation of the phosphoric acid is not completed in presence of ammonium citrate except it is employed in conjunction with a sufficient excess of magnesia. But the foreign matters which accompany the phosphoric acid require different quantities of ammonium citrate in order to keep them in solution, and it is important to increase the magnesium solution at the time of increasing the citric acid in order to maintain them always in the same proportion. This is easily accomplished by measuring the two solutions, but it is much more easily done by uniting them and adding them together.
90. The Magnesium Citrate Solution.—The formula originally proposed by Joulie, and modified by Millot, and adopted by the French Association of Chemists, is as follows: Citric acid, 400 grams; pure magnesium carbonate, forty grams; caustic magnesia, twenty grams; distilled water, half a liter. After solution, add enough of ammonia to render strongly alkaline, requiring about 600 cubic centimeters. Make the volume up with distilled water to one and a half liters. If the solution be turbid, it is proof that the magnesia or the carbonate employed contains some phosphoric acid which is to be separated by filtration, and the solution can then be preserved indefinitely.
91. Time of Subsidence.—When the phosphoric acid is precipitated by the mixture above mentioned, it is necessary to allow it to subside for a certain time under a bell jar in order to avoid the evaporation of the ammonia. In order to give plenty of time for this subsidence, it is well to make the precipitations in the afternoon and the filtrations the following morning. There are thus secured twelve to fifteen hours of repose, which is time amply sufficient for all cases.
92. Filtration and Washing.—Filtration is performed easily and rapidly upon a small filter without folds placed in a funnel with a long [Pg 93] stem of about two millimeters internal diameter. Placed in a series of six or eight, they allow the filtration to take place in regular order without loss of time, the first filter being always empty by the time the last one is filled. The supernatant liquid from the precipitate should first be decanted on the filter, avoiding the throwing of the filtrate on the filter which would greatly retard the process, especially if it should contain a little silica, as often happens.
When the clear liquid is thus decanted as completely as possible, the rest of the precipitate is treated with water to which one-tenth of its volume of ammonia has been added, and the washing is continued by decantation as at first, and afterwards by washing upon the filter until the filtered solution gives no precipitate with sodium phosphate. Four washings are generally sufficient to attain this result.
If the operations which precede have been well-conducted, the total phosphoric acid contained in the material under examination is found upon the filter-paper, except the small portion which remains adhering to the beaker in which the precipitation has been made. The determination of the phosphoric acid comprises the following operations: First, solution of the ammonium magnesium phosphate and second, titration by means of a standard solution of uranium.
93. Solution of the Ammonium Magnesium Phosphate.—The phosphate which has been collected upon the filter is dissolved by a ten per cent solution of pure nitric acid. This solution is caused to pass into the beaker in which the precipitation was made in order to dissolve the particles of phosphate which remain adherent to its sides; and this solution is then thrown upon the filter. The filtrate is then received in a flask of about 150 cubic centimeters capacity, marked at seventy-five cubic centimeters. After two or three washings with the acidulated water, the filter itself is detached from the funnel and introduced into the vessel which contains the solution.
The whole of the filtrate being collected in the flask it is saturated by one-tenth ammoniacal water until a slight turbidity is produced. One or two drops of dilute nitric acid are now added until the liquor [Pg 94] becomes limpid, and the flask is placed upon a sand-bath in order to carry the liquid to the boiling-point. After ebullition there are added five cubic centimeters of acid sodium acetate in order to cause the free nitric acid to disappear and immediately the titration, by means of a standard solution of uranium, is undertaken.
94. Acid Sodium Acetate.—The acid sodium acetate is prepared as follows: Crystallized sodium acetate, 100 grams; glacial acetic acid, fifty cubic centimeters; distilled water, enough to make one liter.
95. Standard Solution of Uranium.—A solution of uranium is to be prepared as follows: Pure uranium nitrate, forty grams; distilled water, about 800 cubic centimeters. Dissolve the uranium nitrate in the distilled water and add a few drops of ammonia until a slight turbidity is produced, and then a sufficient amount of acetic acid to cause this turbidity to disappear. The volume is then completed to one liter with distilled water.
The uranium nitrate often contains some uranium phosphate and some ferric nitrate. It is important that it be freed from these foreign substances. This is secured by dissolving it in distilled water and precipitating it by sodium carbonate, which redissolves the uranium oxid and precipitates the iron phosphate and oxid.
The filtered liquor is saturated with nitric acid, and the uranium oxid reprecipitated by ammonia. It is then washed with distilled water by decantation and redissolved in nitric acid, as exactly as possible, evaporated, and crystallized.
The crystals are taken up with ether, which often leaves still a little insoluble matter. The solution is filtered, and the ether evaporated. The salt which remains is perfectly pure. It frequently happens when the uranium nitrate has not been properly purified that the solution prepared as has been indicated above, deposits a light precipitate of phosphate which alters its strength and affords a cause of error. Only those solutions should be employed which have been prepared some days in advance, and which have remained perfectly limpid. [Pg 95]
The solution of uranium thus obtained contains uranium nitrate, a little ammonium nitrate, a very small quantity of uranium acetate, some ammonium acetate, and a little free acetic acid. Its sensibility is the more pronounced as the acetates present in it are less in quantity. It is important, therefore, never to prepare the solution with uranium acetate.
96. Typical Solution of Phosphoric Acid.—In order to titrate a solution of uranium, it is necessary to have a standard solution of phosphoric acid; that is to say, a solution containing a precise and known quantity of that acid in a given volume. This solution is prepared by means of acid ammonium phosphate, a salt which is easily obtained pure and dry. Sometimes as it may contain a small quantity of neutral phosphate which modifies the relative proportions of phosphoric acid and ammonia, and it is indispensable to have its strength verified. The titer of the typical solution should be such that it requires for the precipitation of the phosphoric acid which it contains, a volume of the solution of uranium almost exactly equal to its own, in order that the expansions or contractions which the two liquors undergo, by reason of changes in the temperature of the laboratory, should be without influence upon the results.
The solution of uranium prepared as has been indicated above, precipitates almost exactly five milligrams of phosphoric acid per cubic centimeter; the typical solution of phosphoric acid is prepared with eight and one-tenth grams of acid ammonium phosphate pure and dry, which is dissolved in a sufficient quantity of distilled water to make one liter.
The acid ammonium phosphate containing 61.74 per cent of anhydrous phosphoric acid, the quantity above gives exactly five grams of that acid in a liter, or five milligrams in a cubic centimeter.
97. Verification of the Strength of the Standard Solution of Phosphoric Acid.—The strength of the standard solution of phosphoric acid is verified by evaporating a known volume, fifty cubic centimeters for example, with a solution of ferric hydroxid containing a known quantity of ferric oxid. The mass having been evaporated to dryness, and ignited in a platinum crucible, gives an increase in the weight of the iron oxid exactly equal to the amount of anhydrous phosphoric acid contained therein, both the nitric acid and ammonia being driven off by the heat. [Pg 96]
To prepare the solution of ferric hydroxid, dissolve twenty grams of iron filings in hydrochloric acid. The solution is filtered to separate the carbon, and it is converted into ferric nitrate by nitric acid, and the solution diluted with distilled water, and the ferric oxid precipitated by a slight excess of ammonia. The precipitate, washed by decantation with distilled water until the wash-water no longer gives a precipitate with silver nitrate, is redissolved in nitric acid, and the solution is concentrated or diluted, as the case may be, to bring the volume to one liter.
In order to determine the quantity of ferric oxid which it contains, fifty cubic centimeters are evaporated to dryness, ignited, and weighed.
A second operation like the above is carried on by adding fifty cubic centimeters of the standard solution of phosphoric acid, and the strength of the solution thus obtained is marked upon the flask.
If the operation have been properly carried on, three or four duplicates will give exactly the same figures. If there are sensible differences, the whole operation should be done over from the first.
98. Titration of the Solution of Uranium.—In a 150 cubic centimeter flask marked at seventy-five cubic centimeters, are poured ten cubic centimeters of the standard solution of phosphoric acid measured with an exact pipette; five cubic centimeters of the acid sodium acetate are added, and distilled water enough to make about thirty cubic centimeters, and the whole carried to the boiling-point. The titration is then carried on by allowing the solution of uranium to fall into the flask from a graduated burette, thoroughly shaking after each addition of the uranium, and trying a drop of the liquor with an equal quantity of a ten per cent solution of potassium ferrocyanid upon a greased white plate. Since the quantity of the uranium solution present will be very nearly ten cubic centimeters at first, nine cubic centimeters can be run in without testing. Afterwards, the operation is continued by adding two or three drops at a time until the test upon the white plate with the potassium ferrocyanid shows the end of the reaction. When there is observed in the final test a slight change of [Pg 97] tint, the flask is filled up to the mark with boiling distilled water and the process tried anew. If in the first part of the operation the point of saturation have not been passed, it is still usually necessary to add a drop or two of the uranium solution in order to produce the characteristic reddish coloration, and this increase is rendered necessary by the increase in the volume of the liquid. Proceeding in this manner two or three times allows the attainment of extreme precision, inasmuch as the analyst knows just when to look for the point of saturation.
Correction.—The result of the preceding operation is not absolutely exact. It is evident indeed that in addition to the quantity of uranium necessary for the exact precipitation of the phosphoric acid, it has been necessary to add an excess sufficient to produce the reaction upon the potassium ferrocyanid.
This excess is rendered constant by the precaution of operating always upon the same volume; namely, seventy-five cubic centimeters. It can be determined then once for all by making a blank determination under the same conditions but without using the phosphoric acid.
The result of this determination is that it renders possible the correction which it is necessary to make by subtracting the quantity used in the blank titration from the preceding result in order to obtain the exact strength of the uranium solution.
The operation is carried on as follows: In a flat-bottomed flask of about 150 cubic centimeters capacity and marked at seventy-five cubic centimeters, by means of a pipette, are placed five cubic centimeters of the solution of sodium acetate; some hot distilled water is added until the flask is filled to the mark, and it is then placed upon a sand-bath and heated to the boiling-point. It is taken from the fire, the volume made up to seventy-five cubic centimeters with a little hot distilled water, and one or two drops of the solution of uranium are allowed to flow into the flask from a graduated burette previously filled exactly to zero. After each drop of the solution of uranium the flask is shaken and the liquid tried upon a drop of potassium ferrocyanid, as has been previously indicated. For a skilled eye, four to six drops are generally necessary to obtain the characteristic coloration; that is from two-tenths to three-tenths of a cubic [Pg 98] centimeter. Beginners often use from five-tenths to six-tenths, and sometimes even more.
The sole important point is to arrest the operation as soon as the reddish tint is surely seen, for afterwards the intensity of the coloration does not increase proportionally to the quantity of liquor employed.
It is well to note that at the end of some time the coloration becomes more intense than at the moment when the solutions are mixed, so that care must be taken not to pass the saturation-point. This slowness of the reaction is the more marked as there is more sodium or ammonium acetate in the standard solutions. This is the reason that it is important to introduce always the same quantity; namely, five cubic centimeters. This is also the reason why the uranium acetate should not be employed in preparing the standard solution of uranium which ought to contain the least possible amount of acetate in order that the necessary quantity which is carried into each test should be as small as possible and remain without appreciable influence. If it were otherwise, the sensibility of the reaction would be diminished in proportion as a larger quantity of uranium solution was employed, giving rise to errors which would be as much more important as the quantities of phosphoric acid to be determined were greater. The correction for the uranium solution having been determined it is written upon the label of the bottle containing it.
Causes of Errors.—In the work which has just been described, some causes of error may occur to which the attention of analysts should be called.
The first is the error which may arise from the consumption of the small quantity of uranium phosphate which is taken with a stirring rod when the liquid is tested with potassium ferrocyanid. It is very easy to be assured that the end of the reaction has really been reached. For this purpose it is only necessary to note the quantity of the solution already employed and to add to it afterwards four drops; shake, and make a new test with a drop of the potassium ferrocyanid placed near the spot which the last one occupied. If a decidedly reddish tint does not appear at the moment of removing the glass rod, it is to be [Pg 99] concluded that the first appearance was an illusion, and the addition of uranium is to be continued. If, on the contrary, the coloration appear of a decided tint, the preceding number may be taken for exact. It is then always beneficial to close the titration by this test of four supplementary drops which will exaggerate the coloration and confirm the figure found.
The second cause of error, and one moreover which is the most frequently met with, consists in passing the end of the reaction by adding the uranium too rapidly. In place of giving then a coloration scarcely perceptible, the test with the drop of potassium ferrocyanid gives a very marked coloration. In this case the analysis can still be saved. For this purpose the analyst has, at his disposal, a tenth normal solution prepared with 100 cubic centimeters of the standard solution of phosphoric acid diluted to one liter with distilled water. Ten cubic centimeters of this tenth normal solution are added, and the titration continued. At the end, the amount of additional phosphoric acid used is subtracted from the total.
A third cause of error is found in the foam which is often found in the liquid, due to the shaking. This foam may retain a portion of the last drops of the solution of uranium which fall upon its surface and prevent its mixture with the rest of the liquid. If the glass stirring rod in being removed from the vessel pass through this froth charged with uranium, the characteristic coloration is obtained before real saturation is reached. Consequently it is necessary to avoid, as much as possible, the formation of the foam, and especially to take care never to take the drop for test after agitation except in the middle of the liquid where the foam does not exist.
Suppose the titration has been made upon ten cubic centimeters of the normal solution of phosphoric acid in the conditions which we have just indicated, and the figure for the uranium obtained is 10.2 cubic centimeters; if now the correction, which may be supposed to amount to two-tenths cubic centimeter, be subtracted there will remain ten cubic centimeters of the uranium solution which would have precipitated exactly fifty milligrams of phosphoric acid. [Pg 100]
The quantity of phosphoric acid which precipitates one cubic centimeter of the solution will be consequently expressed by the proportion ⁵⁰/₁₀ = five milligrams, which is exactly the strength required. In the example which has just been given, the inscription upon the flask holding the standard solution would be as follows: Solution of uranium, one cubic centimeter equals five milligrams of phosphorus pentoxid; correction, two-tenths cubic centimeter.
99. Titration of the Sample.—The strength of the solution of uranium having been exactly determined, by means of this solution the strength of the sample in which the phosphoric acid has been previously prepared as ammonium magnesium phosphate is ascertained. In this case the quantity of phosphoric acid being unknown, it is necessary to proceed slowly and to duplicate the tests in order not to pass beyond the point of saturation. From this there necessarily results a certain error in consequence of the removal of quite a number of drops of the solution of the sample before the saturation is complete. It is therefore necessary to make a second determination in which there is at once added almost the quantity of the solution of uranium determined by the first analysis. Afterwards the analysis is finished by additions of very small quantities of uranium until saturation is reached. Suppose, for instance, that the sample was that of a mineral phosphate, five grams of which were dissolved in 100 cubic centimeters, and of which ten cubic centimeters of the solution prepared as above required 15.3 cubic centimeters of the standard solution of uranium. We then would have the following data:
Mineral phosphate, five grams of the material dissolved in twenty cubic centimeters of hydrochloric acid.
Water, sufficient quantity to make 100 cubic centimeters.
Quantity taken, ten cubic centimeters = 0.50 gram of the sample taken.
| Solution of uranium required | 15.3 | cubic | centimeters. |
| Correction | 0.2 | “ | “ |
| Actual quantity of uranium solution | 15.1 | “ | “ |
Strength of the solution of uranium, one cubic centimeter = fivemilligrams P₂O₅.
Then P₂O₅ in 0.50 gram of the material = 5 × 15.1 = 75.50 milligrams.
| Then the per cent of P₂O₅ = | 75.5 x 100 | = 15.10. |
| 50 |
[Pg 101] The sample under examination ought always to be prepared in duplicate, either by making a single precipitation and re-solution of the ammonium magnesium phosphate which is made up to a certain volume and an aliquot portion of which is taken for the analysis, or by making two precipitations under the conditions previously described. When the content of phosphoric acid in the material under examination is very nearly known, the double operation may be avoided, especially if it be required to have rapid and only approximate analyses, such as those which are made for general control and for the conduct of manufacturing operations. But when analyses are to be used to serve as the basis of a law or for the control of a market, they should always be made in duplicate, and the results ought not to be accepted when the numbers obtained are widely different, since the agreement of the two numbers will show that the work has been well executed.
This method of analysis, much longer to describe than to execute, gives results perfectly exact and always concordant when it is well carried out, provided that the standard solutions, upon which it rests for its accuracy, are correctly prepared and frequently verified in the manner indicated.
The strength of the solution of uranium ought to be verified every three or four days. The strength of the standard solution of phosphoric acid should be verified each time that the temperature of the laboratory undergoes any important change. A solution prepared, for example, in winter when the temperature of the laboratory is from 15° to 18° would no longer be exact in summer when the temperature reaches 28° or 30°.
100. Condition of Phosphoric Acid in Superphosphates.—Superphosphates are the products of the decomposition of phosphates by sulfuric or hydrochloric acid. They contain phosphoric acid combined with water, with lime, with magnesia, and with iron and alumina in various proportions. [Pg 102]
These combinations may be classed in three categories: First, those compounds soluble in water; second, those insoluble in water, but very soluble in ammoniacal salts of the organic acids such as the citrate and oxalate; and third, phosphates not soluble in any of the above-named reagents.
In the products soluble in water are met free phosphoric acid, monocalcium phosphate, acid magnesium phosphate, and the iron and aluminum phosphates dissolved in the excess of phosphoric acid. In the products insoluble in water but soluble in the ammonium citrate are found bicalcium phosphate and iron and aluminum phosphates, which together constitute the phosphates called reverted.
These compounds reduced to a very fine state of division in the process of manufacture are considered to contain phosphoric acid of the same economic value.
101. Determination of the Total Phosphoric Acid in Superphosphates and Fertilizers.—The process is carried on exactly as for an ordinary phosphate, and with all the care indicated in connection with the sampling, the incineration, the solution by means of hydrochloric acid, and the separation of the phosphoric acid in the state of ammonium magnesium phosphate, and finally in the titration by uranium.
102. Determination of Soluble and Reverted Phosphoric Acid.—To make this determination a method unique and applicable to all cases consists in extracting, at first, the soluble constituents in distilled water, and following this operation by digestion in the ammonium citrate. The products soluble in water can be determined either separately or at the same time as the products soluble in the ammonium citrate according to the taste of the people interested, without its being necessary to modify very greatly the method of operation.
The determination of the soluble phosphoric acid comprises first, the solution of the soluble constituents in distilled water; second, the solution of the reverted phosphates in ammonium citrate; third, the precipitation of the phosphoric acid dissolved in the two preceding operations, and its determination.
103. Preparation of the Sample for Analysis.—The sample sent to the chemical expert is prepared as has been indicated; that is to say, it is poured on a sieve of which the meshes have a diameter of one [Pg 103] millimeter, and sifted upon a sheet of white paper. The parts which do not pass the sieve are broken up either by the hand or in a mortar and added, through the sieve, to the first portions. The product is well mixed and, in this state, the mass presents all the homogeneity desirable for analysis.
Some fertilizers are received in a pasty state which does not permit of their being sifted. It is necessary in such a case to mix them with their own weight either of precipitated calcium sulfate dried at 160° or with fine sand washed with hydrochloric acid and dried, which divides the particles perfectly and permits of their being passed through the meshes of the sieve.
104. Extraction of the Products Soluble in Distilled Water.—The substance having been prepared as has just been indicated, one and a half grams are placed in a glass mortar. Twenty cubic centimeters of distilled water are added, and the substance gently suspended therein. After standing for one minute, the supernatant part is decanted into a small funnel provided with a filter-paper and placed in a flask marked at 150 cubic centimeters. This operation is repeated five times and is terminated by an intimate breaking up of the matter with distilled water. When the volume of 100 cubic centimeters of the filtrate has been obtained, the residue in the mortar is placed on the filter, and the washing is continued until the total volume reaches 150 cubic centimeters. The filtrate is shaken in order to render the liquor homogeneous, and is transferred to a precipitating glass of about 300 cubic centimeters capacity.
105. Solution of the Reverted Phosphates by Ammonium Citrate.—The filter from the above process is detached from the funnel and is introduced into a flask marked at 150 cubic centimeters together with sixty cubic centimeters of alkaline ammonium citrate prepared in the following manner:
The ammonia is poured upon the citric acid in the form of crystals in a large dish. The mass becomes heated, and the solution takes place rapidly. When it is complete and the solution is cold it is poured into a flask of one liter capacity, and the flask is filled up to the mark [Pg 104] with strong ammonia. It is preserved for use in a well-stoppered bottle. The solution must be strongly alkaline.
The flask in which the filter-paper is introduced, together with the ammonium citrate, is stoppered and shaken violently in order to disintegrate the filter-paper and put the reverted phosphates in suspension. There are added then about sixty cubic centimeters of distilled water, and the flask is shaken and left for twelve hours at least, or at most for twenty-four hours. The volume is made up to 150 cubic centimeters with distilled water, and, after mixture, the solution is filtered.
There are thus obtained two solutions which can be precipitated together or separately according to circumstances. The most usual process is to combine the two equal volumes of twenty-five, fifty, or one hundred cubic centimeters, representing one-quarter, one-half, or one gram of the material according to its presumed richness, in a precipitating flask to which are added from ten to twenty cubic centimeters of the solution of magnesia made up as follows:
| Magnesium carbonate, | 50 | grams. | |
| Ammonium chlorid, | 100 | “ | |
| Water, | 500 | cubic | centimeters. |
| Hydrochloric acid, | 120 | “ | “ |
After complete solution of the solid matters in the above, add 100 cubic centimeters of ammonia of 22° strength, and distilled water enough to make one liter.
The solutions are thoroughly mixed in a precipitating glass, an excess of ammonia added, and allowed to stand for twelve hours under a bell jar. The phosphoric acid contained in the liquor is separated as ammonium magnesium phosphate. It is collected upon a small filter, washed with a little ammoniacal water, redissolved, and titrated with the uranium solution in the manner already indicated.
Example: The following is an example of this kind of a determination:
(1) One and one-half grams of the superphosphate and distilled water enough to make 150 cubic centimeters. [Pg 105]
(2) Filter-paper with reverted phosphates, sixty cubic centimeters of ammonium citrate, and a sufficient quantity of distilled water to make 150 cubic centimeters.
| Aqueous solution | (1) | 25 cc | = 0.25 grams of the sample. | |
| Citrate solution | (2) | 25 cc |
Add magnesium solution twenty cubic centimeters and ammonia in excess, and allow from twelve to twenty-four hours of digestion, then filter and wash, dissolve and titrate.
Required of solution of uranium 8.55 cubic centimeters (1 cubic centimeter = 5 milligrams P₂O₅).
Correction 0.20.
Remainder 8.35 × 0.005 = 0.04175 gram P₂O₅ for 0.25 gram of the sample. Then 0.04175 ÷ 0.25 = 16.7 per cent.
From the above data there would be 16.7 per cent of phosphoric acid soluble in water and in ammonium citrate.
If it be desirable to have separately the phosphoric acid soluble in water, a separate precipitation is made of the aqueous solution alone by means of the magnesium citrate solution. The precipitate washed with ammoniacal water is redissolved and titrated in the manner indicated.
In subtracting from the figures obtained with the two solutions together the number obtained for the phosphoric acid soluble in water, the number representing the phosphoric acid soluble in ammonium citrate alone, is obtained.
It is to be noted that the determinations with uranium require always two successive titrations. It would therefore be an advantage in all operations to precipitate a weight of ammonium magnesium phosphate sufficient for allowing this precipitate to be dissolved and made up to 100 cubic centimeters on which amount it would be possible to execute two, three, or four determinations, and thus to obtain a figure absolutely incontestable.
106. Conclusions.—It has been seen from the above data that the French chemists have worked out the uranium volumetric method with great patience and attention to detail. Where many determinations are to be made it is undoubtedly possible for an analyst to reach a high degree of accuracy as well as to attain a desirable rapidity, by using this method. For a few determinations, however, the labor of preparing and setting the standard solutions required would be far greater than [Pg 106] the actual determinations either by the molybdate or citrate gravimetric methods. For control work in factories and for routine work connected with fertilizer inspection, the method has sufficient merit to justify a comparison with the processes already in use by the official chemists of this country.
The use of an alkaline ammoniacal citrate solution, however, for the determination of reverted acid renders any comparison of the French method with our own impossible. On the other hand the French method for water-soluble acid is based on the same principle as our own; viz., washing at first with successive small portions of water, and thus avoiding the decomposition of the soluble phosphates, which is, likely to occur when too great a volume of water is added at once.
In the matter of the temperature and time as affecting the solubility of reverted acid, the French method is also distinctly inferior to our own. The digestion is allowed to continue from twelve to twenty-four hours, at the pleasure of the analyst, and meanwhile it is subjected to room temperature. It is not difficult to see that this treatment in the same sample would easily yield disagreeing results between twelve hours at a winter temperature and twenty-four hours at summer heat.
107. Pemberton’s Volumetric Method.—In order to shorten the work of determining the phosphoric acid, numerous attempts have been made to execute the final determination directly on the yellow precipitate obtained by treating a solution of a phosphate with ammonium molybdate in nitric acid. The composition of this precipitate appears to be somewhat variable, and this fact has cast doubt on the methods of determination based on its weight. Its most probable composition is expressed by the following formula, (NH₄)₃PO₄(MoO₃)₁₂. For convenience in writing reactions this formula should usually be doubled. Pemberton has described a volumetric determination of phosphoric acid in the yellow precipitate which has the merit of being rapid.[83]
[Pg 107] In this laboratory the method has not given very satisfactory results when compared with the molybdate gravimetric process. It has however attracted so much attention from analysts as to merit description, and the details of the process are therefore given.
108. The Process.—One gram of phosphate rock, or from two to three grams of phosphatic fertilizer, are dissolved in nitric acid, and, without evaporation, diluted to 250 cubic centimeters. Without filtering, twenty-five cubic centimeters are placed in a four-ounce beaker and ammonia added until a slight precipitate begins to form. Five cubic centimeters of nitric acid of one and four-tenths specific gravity are then added, and afterwards ten cubic centimeters of saturated solution of ammonium nitrate and enough water to make the volume about sixty-five cubic centimeters. The contents of the beaker are boiled, and while still hot, five cubic centimeters of the aqueous solution of ammonium molybdate added. Additional quantities of the molybdate are added, if necessary, until the whole of the phosphorus pentoxid is thrown-out.
After allowing to settle for a moment the contents of the beaker are poured upon a filter seven centimeters in diameter. The precipitate is thoroughly washed with water, both by decantation and on the filter. The filter with its precipitate is transferred to a beaker and titrated with standard alkali, in the presence of phenolphthalein. Each cubic centimeter of alkali employed should correspond to one milligram of phosphorus pentoxid, (P₂O₅).
The reagents employed have the composition indicated below:
Ammonium Molybdate.—Ninety grams of the crystals of ammonium molybdate are placed in a large beaker and dissolved in a little less than one liter of water. The beaker is allowed to stand over night and the clear liquor decanted. Any undissolved acid is brought into solution in a little ammonia water and added to the clear liquor. If a trace of phosphoric acid be present a little magnesium sulfate is added and enough ammonia to produce a slight alkaline reaction. The volume of the solution is then made up to one liter. Each cubic centimeter of this solution is capable of precipitating three milligrams of phosphorus pentoxid.
Standard Potassium Hydroxid.—This solution is made of such strength that one cubic centimeter is equivalent to one milligram of phosphorus pentoxid. Treated with acid of normal strength, 100 cubic [Pg 108] centimeters are required to neutralize 32.37 cubic centimeters thereof.
Standard Acid.—This should have the same strength, volume for volume, as the standard alkali solution. It is made by diluting 323.7 cubic centimeters of normal acid to one liter.
Indicator.—The indicator to be used is an alcoholic solution of phenolphthalein, one gram in 100 cubic centimeters of sixty per cent alcohol, and half a cubic centimeter of this should be used for each titration.
Thomson has shown[84] that of the three hydrogen atoms in phosphoric acid two must be saturated with alkali before the reaction with phenolphthalein is neutral. Therefore, when the yellow precipitate is broken up by an alkali, according to the reaction to follow, only four of the six molecules of ammonium are required to form a neutral ammonium phosphate as determined by the indicator employed. The remaining two molecules of ammonium unite with the molybdenum forming also a salt neutral to the indicator.
Phenolphthalein is preferred because, as has been shown by Long, its results are reliable in the presence of ammonium salts unless they be present in large quantity, and if the solution be cold and the indicator be used in sufficient quantity.[85] To prepare the indicator for this work, one gram of phenolphthalein is dissolved in 100 cubic centimeters of sixty percent alcohol. At least one-half of a cubic centimeter of the solution is used for each titration.
The advantages claimed for the method are its speed and accuracy. Much time is saved by avoiding the necessity for the removal of the silica by evaporation. The results of analyses with and without the removal of the silica are practically identical. When the silica is not removed it is noticed that the filtrate from the yellow precipitate has a yellow tint.
The reaction is represented by the following formula:
(NH₄)₆(PO₄)₂(MoO₃)₂₄ + 46KOH = (NH₄)₄(HPO₄)₂ + (NH₄)₂MoO₄
+ 23K₂MoO₄ + 22H₂O.
From this reaction it is seen that the total available acidity of one molecule of the yellow precipitate titrated against phenolphthalein is equivalent to twenty-three molecules of potassium hydroxid. [Pg 109]
Calculation of Results.—The standard alkali is of such strength that one cubic centimeter is equal to one per cent of phosphoric acid when one gram of material is employed and one-tenth of it taken for each determination. In a given case one gram of a sample was taken and one-tenth of the solution used. Fifty cubic centimeters of alkali were added to the yellow precipitate. It required thirty-two cubic centimeters of standard alkali to neutralize the excess.
The alkali consumed by the yellow precipitate was 50 - 32 = 18. The sample therefore contained eighteen per cent of phosphoric acid.
Comparison with Official Method.—A comparison of the Pemberton volumetric with the official method of the Association of Agricultural Chemists has been made by Day and Bryant.[86] The comparisons were made on samples containing from 1.45 to 37.28 per cent of phosphoric acid and resulted as follows:
| Substance. | Per cent P₂O₅, Official. |
Per cent P₂O₅, Pemberton. |
||
|---|---|---|---|---|
| No. 1. | Florida | rock | 1.45 | 1.32 |
| “ 2. | “ | “ | 4.40 | 4.53 |
| “ 3. | Sodium | phosphate | 19.78 | 19.99 |
| “ 4. | “ | “ | 19.72 | 19.73 |
| “ 5. | Florida | rock | 37.28 | 37.22 |
This near agreement shows the reliability of the method. The comparison of the Pemberton volumetric method with the official gravimetric method was investigated by the reporter of the Association of Official Agricultural Chemists in 1894.[87] The individual variations were found to be greater than in the regular method but the average results were nearly identical therewith. The method works far better with small percentages of phosphoric acid than with large. Where the average of the results by the official methods gave 12.25 per cent, the volumetric process gave 11.90 per cent, whereas in the determination of a smaller percentage the results were 2.72 and 2.73 per cent, respectively. Kilgore proposes a variation of the method which differs from the original in two principal points.[88] First the temperature of precipitation in the Pemberton process is 100°; but in the modified form from 55° to 60°. At the higher temperature there is danger of depositing molybdic acid.
The second difference is in the composition of the molybdate solution employed. The official molybdate solution contains about sixty grams [Pg 110] of molybdenum trioxid in a liter while the Pemberton solution contains sixty-six grams. There is therefore not much difference in strength. The absence of nitric acid, however, from the Pemberton solution favors the deposition of the molybdic acid when heat is applied. Kilgore, therefore, conducts the analysis as follows: The solution of the sample is made according to the official nitric and hydrochloric acid method for total phosphoric acid. For the determination, twenty or forty cubic centimeters are taken, corresponding to two-tenths or four-tenths gram of the sample. Ammonia is added until a slight precipitate is produced and the volume is then made up, with water, to seventy-five cubic centimeters. Add some ammonium nitrate solution, from ten to fifteen cubic centimeters, but this addition is not necessary unless much of the nitric acid has been driven off during solution. Heat in water-bath to 60° and precipitate with some freshly filtered official molybdate solution. Allow to stand for five minutes, filter as quickly as possible, wash four times by decantation using from fifty to seventy-five cubic centimeters of water each time, and then wash on a filter until all acid is removed. The solution and titration of the yellow precipitate are accomplished as in the Pemberton method. The agreement of the results obtained by this modified method was much closer with the official gravimetric method than those obtained by the Pemberton process.
109. Estimation of Phosphoric Acid as a Lead Compound.—In the volumetric lead method, as described by Wavelet, the phosphoric acid is precipitated by the magnesium citrate solution as in the uranium method of Joulie, as practiced by the French chemists, and the washing of the precipitate and its solution in nitric acid are also conducted as in that method.[89] After solution in nitric acid ammonia is added to neutrality and the solution is then made acid with acetic. The phosphoric acid is precipitated in the acid solution by a standard solution of lead nitrate, the precipitate having the formula P₂O₅3PbO.
The end reaction is determined by placing a drop of the titrated mixture on a white greased dish in contact with a drop of a five per cent solution of potassium iodid. When all the phosphoric acid is precipitated the least excess of the lead salt is revealed by the characteristic yellow precipitate of lead iodid. [Pg 111]
The author of the process claims that the lead phosphate is insoluble in the excess of acetic acid and that the phosphate itself does not give any yellow coloration with potassium iodid. The process is quite as exact as the uranium method and the end reaction is far sharper and the standard reagents are easily made and preserved.[90] The method described merits, at least, a comparative trial with the uranium process, but cannot be recommended as exact until further approved by experience.
The reagents employed have the following composition:
| (1) | Disodium phosphate | solution | containing | 10.085 | grams | per | liter |
| (2) | Sodium acetate | “ | “ | 50.000 | “ | “ | “ |
| (3) | Lead nitrate | “ | “ | 40.000 | “ | “ | “ |
| (4) | Potassium iodid | “ | “ | 50.000 | “ | “ | “ |
| The titrations should be conducted in the cold. | |||||||
110. Water-Soluble Phosphoric Acid.—Glaser has modified the volumetric method of Kalmann and Meissels for the volumetric estimation of water-soluble phosphoric acid so as to avoid the double titration required by the original method.[91] If methyl orange be used as an indicator in the original method, the determination does not at once lead to the tricalcium salt, but the liquid still contains, after neutralization, some monocalcium phosphate, which is determined by a further titration with phenolphthalein. In the modified method the total phosphoric acid is estimated in one operation as a tricalcium salt. This is secured, by adding, at the proper time, an excess of calcium chlorid. Two grams of the superphosphate are shaken with water several times, and, after settling, filtered, and the insoluble residue finally washed on the filter until the total volume of the filtrate is a quarter of a liter. Of this, fifty cubic centimeters are taken and titrated with tenth normal soda-lye, with addition of two drops of methyl orange, until the acid reaction has entirely disappeared. There is then added some neutral calcium chlorid solution in excess. If iron and alumina be present, a precipitate is produced of which no account need be made. The acid reaction is thus restored. Five drops of the phenolphthalein solution are added and the titration continued until the alkaline reaction is noted throughout the whole mass. Each cubic centimeter of the soda-lye corresponds, in the first titration, to 7.1, and in the second to 3.55 milligrams of phosphoric acid. [Pg 112]
111. Estimation of Phosphoric Acid in the Presence of a Large Excess of Iron.—The method given below, due to Emmerton, depends upon the precipitation of a phosphomolybdate, of constant composition, in the presence of a large excess of iron, as in the analysis of iron and steel and iron ores.[92] The molybdenum trioxid obtained is reduced by zinc to Mo₁₂O₁₉. The action of permanganate on this compound is shown in the following equation:
5Mo₁₂O₁₉ + 17(K₂OMn₂O₇) = 60MoO₃ + 17K₂O + 34MnO.
Seventeen molecules of permanganate are equal to sixty molecules of molybdenum trioxid. The iron or steel is dissolved in nitric acid, evaporated to dryness, heated, and redissolved in hydrochloric acid, then treated again with nitric acid and evaporated until a clear and concentrated solution is obtained free from hydrochloric acid.
The solution obtained is diluted to forty cubic centimeters with water and washed into a 400 cubic centimeter flask, making the total volume about seventy-five cubic centimeters. Add strong ammonia, shaking after each addition, until the mass sets to a thick jelly from the ferric hydroxid. Add a few more cubic centimeters of ammonia and shake thoroughly, being sure the ammonia is present in excess. Add next nitric acid gradually, with shaking, until the precipitate has all dissolved; add enough more nitric acid to make the solution a clear amber color. The volume should now be about 250 cubic centimeters. Bring the solution to 85° and add, at once, forty cubic centimeters of molybdate solution of the following strength: Dissolve 100 grams of molybdic acid in 300 cubic centimeters of strong ammonia and 100 cubic centimeters of water, and pour the solution into 1,250 cubic centimeters of nitric acid (1.20); close the flask with a rubber stopper, wrap it in a thick cloth, and shake violently for five minutes. Collect the precipitate on a filter, using pump, and wash with dilute nitric acid (1 HNO₃ : 50 H₂O). If a thin film of the precipitate should adhere to the flask it can be removed by the ammonia in the next operation. Wash the molybdate precipitate into a 500 cubic centimeter flask with dilute ammonia (1 H₃N : 4 H₂O), using about thirty cubic centimeters. Add hot dilute sulfuric acid (1 H₂SO₄ : 4 H₂O) and cover the flask with a small funnel. Add ten grams of granulated zinc and heat [Pg 113] until rapid action begins, and then heat gently for five minutes. The reduction is then complete. During the reduction the colors, pink, plum, pale green, and dark green, are seen in the molybdate solution, the latter color marking the end of the reaction.
To remove the zinc, pour through a large folded filter, wash with cold water, and fill up the filter once with cold water. But little oxidation takes place in this way. A port-wine color is seen on the filter, but this does not indicate a sufficient oxidation to make an error.
In titrating, the wine color becomes fainter and finally the solution is perfectly colorless and shows a single drop in excess of the permanganate. The permanganate solution, for convenience, is made so that one cubic centimeter is equal to 0.0001 gram of phosphorus. With iron its value is one cubic centimeter equals 0.006141 gram of iron; and one cubic centimeter equals 0.005574 gram of molybdenum trioxid.
112. Variation of Dudley and Noyes.—The method of Emmerton to determine small quantities of phosphoric acid, or of phosphorus in presence of a large excess of iron, has been modified by Dudley and Pease,[93] and by Noyes and Royse.[94] As modified, the method is not intended for fertilizer analysis, but the principle on which it rests may some time, with proper modifications, find application in fertilizer work. The reduction is accomplished in a Jones’ tube,[95] much simplified, so as to render it suitable for common use. The molybdic acid is reduced to a form, or series of forms, corresponding to molybdenum sesquioxid, as in the Emmerton method, and subsequently as in that method, titrated by a set solution of potassium permanganate.
The iron or steel filings, containing phosphorus, are brought into solution by means of nitric acid. For this purpose two grams of them are placed in a half liter flask together with fifty cubic centimeters of nitric acid of 1.18 specific gravity. The mixture is boiled for one minute, and ten cubic centimeters of permanganate solution of one and a quarter per cent added. Boil again until the pink color disappears. Ferrous sulfate solution is next to be carefully added, shaking meanwhile, until the solution clears. Cool to 50° and add eight cubic centimeters of ammonia of 0.90 specific gravity, stopper the flask, and shake until any precipitate which may form is redissolved. Cool or warm, [Pg 114] as the case may be, until the solution is as many degrees above or below 60° as the molybdic solution is above or below 27°. Add sixty cubic centimeters of molybdic solution, stopper, and shake on a machine or by hand for five minutes. After remaining at rest for five minutes pour into a nine centimeter filter of fine texture and wash with the acid ammonium sulfate solution in quantities of from five to ten cubic centimeters each time. The filtrate and washings must be perfectly bright. Continue the washings until the filtrate gives no color with hydrogen sulfid.
Dissolve the yellow precipitate with twelve cubic centimeters of 0.96 ammonia diluted with an equal volume of water, and wash the filter with 100 cubic centimeters of water. Finally add to the filtrate and wash-water eighty cubic centimeters of water and ten of strong sulfuric acid. Pass the mixture through the Jones’ reducing tube and follow it with 200 cubic centimeters of water, taking care that no air enter the tube during the operations. The solution collected in the flask should be at once titrated with potassium permanganate.
Solutions used: (1) Nitric acid.—One part of nitric acid of 1.42 specific gravity and two parts of water by volume. The specific gravity of the mixture is about 1.18.
(2) Permanganate solution for oxidizing.—Dissolve 12.5 grams of potassium permanganate in one liter of water.
(3) Ferrous sulfate.—Fresh crystals not effervesced and free from phosphorus.
(4) Ammonia.—The strong ammonia used should have a specific gravity of about 0.90 and the dilute of 0.96 at 15.5°.
(5) Molybdic Solution.—Dissolve 100 grams of molybdic anhydrid in 400 cubic centimeters of ammonia of 0.96 specific gravity and pour the solution slowly, with constant stirring, into one liter of nitric acid of about 1.20 specific gravity. Heat the mixture to 45° and add one cubic centimeter of a ten per cent solution of sodium phosphate, stir vigorously, and allow to stand in a warm place for eighteen hours. Filter before using.
(6) Add Ammonium Sulfate.—To half a liter of water add 27.5 cubic centimeters of 0.96 ammonia and twenty-four cubic centimeters of strong sulfuric acid, and make the volume one liter with water. [Pg 115]
(7) Potassium Permanganate for Titration.—Dissolve four grams of potassium permanganate in two liters of water, heat nearly to boiling for an hour, allow to stand for eighteen hours, and filter on asbestos felt. The solution must not come in contact with rubber or other organic matter. The solution may be standardized with thoroughly air-dried ammonium oxalate in solution with a little dilute sulfuric acid and with ammonium ferrous sulfate partly crystallized in small crystals from a slightly acid solution. The crystals should be well washed and quickly air-dried in a thin layer. The factors 1/1 1/4 2/2 and 1/7 should be used respectively to calculate the iron equivalent. The phosphorus equivalent is obtained by multiplying the iron equivalent by 31 ÷ (36 × 56) = 0.01538.
Figure. 7.
Jones’
Reduction
Tube.
Reduction Apparatus.—The reduction of the molybdic acid to molybdenum trioxid is accomplished in a tube first proposed by Jones. The apparatus is shown in Figure 7. A piece of moderately heavy glass tubing thirty-five centimeters long with an internal diameter of two centimeters is drawn out at the lower end so as to pass into the stopper of a flask. A circular piece of perforated platinum or porcelain rests on the constricted portion of the tube and this is covered with an asbestos felt. The tube is then nearly filled with powdered zinc which is washed, before using, with dilute sulfuric acid (1 : 20). A, B, C represent different methods of filtering the molybdic solution. In A a platinum cone is placed in the constricted portion of the tube and the asbestos felt placed thereon and the tube then filled with the granulated zinc. In B there is first inserted a perforated disk then some very fine sand and this is covered with another disk. In C there is a perforated disk which is covered with asbestos felt. The filtering arrangement should be such as to prevent any zinc particles from reaching the flask and yet permitting the filtration to go on without much difficulty. A blank determination is first made by adding to 180 cubic centimeters of water, twelve of 0.96 ammonia and ten of [Pg 116] strong sulfuric acid. This is poured through the reducing tube and followed with 200 cubic centimeters of water taking care that no air enter the apparatus. Hydrogen peroxid is formed if air enter. Even after standing for a few moments the tube should be washed with dilute sulfuric acid before again using it. The filtrate should be titrated with the permanganate solution and the amount required deducted from the following amounts obtained with the molybdic salt.
Calculations.—The calculations of the amount of phosphorus in a given sample of iron or steel are made according to the following data: In a given case let it be supposed that the permanganate solution is set with a solution of piano wire and it is found that one cubic centimeter of permanganate liquor is equal to 0.003466 gram of metallic iron. It is found that 90.76 parts of molybdic acid will produce the same effect on permanganate as 100 parts of iron. Hence one cubic centimeter of permanganate solution is equivalent to 0.003466 × 0.9076 = 0.003145 gram of molybdic acid. In the yellow precipitate formed, in the conditions named for the analysis it is found that the phosphorus is one and nine-tenths per cent of the molybdic acid present. Therefore one cubic centimeter of permanganate liquor is equal to 0.003145 × 0.019 = 0.0000597 gram of phosphorus. If then, for example, in a sample of iron or steel eight and six-tenths cubic centimeters of permanganate solution, after correction, be found necessary to oxidize the molybdic solution after passing through the Jones’ reducing tube, the amount of phosphorus found is 0.0000597 × 8.6 = 0.051 per cent.
113. The Silver Method.—The separation of the phosphoric acid by silver according to the method of Perrot has been investigated by Spencer, who found the process unreliable.[96] By a modification of the process, however, Spencer obtained fairly satisfactory results. The principle of this method depends on the separation of the phosphoric acid by silver carbonate and the subsequent titration thereof with standard uranium solution after the removal of the excess of silver. The operation is conducted as follows: The fertilizer is first ignited until all organic matter and residual carbon are destroyed. Solution is then accomplished by means [Pg 117] of nitric acid and the volume completed to a definite quantity. An aliquot part is taken, after filtration, varying with the supposed strength of the solution so as to contain about 100 milligrams of phosphorus pentoxid. In the slightly nitric acid solution add freshly prepared silver carbonate in excess, that is, sufficient to saturate any free acid present and also to combine with all the phosphoric acid. Wash thoroughly with hot water and then dissolve the mixed phosphate and silver carbonate in nitric acid and remove the silver from the solution with sodium chlorid. The phosphoric acid is determined in the filtrate by means of a standard solution of uranium nitrate in the manner already described. Spencer found that the separation of the phosphoric acid by the silver method was more exact than by the Joulie magnesium citrate process. With practice on the part of the analyst in determining the end reaction the process is both rapid and accurate. The method is also inexpensive, as both the silver and uranium are easily recovered from the waste.
114. Volumetric Silver Method.—Holleman has proposed a modification of the silver method for the volumetric determination of phosphoric acid, which is conducted in the following manner:[97]
In a flask of 200 cubic centimeters capacity, are placed fifty cubic centimeters of the liquid to be analyzed, which should not contain more than two-tenths gram of phosphoric acid. The solution is treated with ten cubic centimeters of a normal solution of sodium acetate and afterwards with a slight excess of decinormal silver solution, four and five-tenths cubic centimeters for each 0.01 gram of phosphoric acid. The solution is then neutralized with tenth normal sodium hydroxid, the amount required having been previously determined by titrating ten cubic centimeters of the liquid to be analyzed, using phenolphthalein as an indicator. Five times the quantity required for the neutralization of the ten cubic centimeters is added, less one-half cubic centimeter. By this treatment the phosphoric acid in the presence of sodium acetate is completely precipitated as silver phosphate. The excess of silver is determined by diluting the mixture to 200 cubic centimeters, filtering, and titrating 100 cubic centimeters of the filtrate with ammonium thiocyanate. The presence of sulfuric and nitric [Pg 118] acids does not interfere with the reaction, but of course hydrochloric acid must be absent. Alkalies and alkaline earth metals may be present, but not the heavy metals.
When iron and aluminum are present 100 cubic centimeters of the solution are precipitated with thirty cubic centimeters of normal sodium acetate, the phosphoric acid is determined in fifty cubic centimeters of the filtrate, and the precipitate of iron and aluminum phosphates is ignited and weighed, and its weight multiplied by 2.225 is added to the phosphoric anhydrid found volumetrically. If ammonia be present it must be removed by boiling, as otherwise it affects the titration with phenolphthalein.
For agricultural purposes this method can have but little value inasmuch as the phosphates to be examined almost always have a certain proportion of iron and aluminum. Inasmuch as the amount of these bases has to be determined gravimetrically, there would be no gain in time and no simplification of the processes by the use of the volumetric method as proposed.
115. Desirability Of Methods.—In the preceding paragraphs, has been given a statement of the principal methods now in use by chemists and others connected with fertilizer control for the scientific and agronomic determinations of phosphoric acid, and its agricultural value.
A résumé of the important methods, in a form suited to use in a factory for preparing phosphatic fertilizers for the market, seems desirable. In these factories the chemists have been accustomed to use their own, or private methods, and there has not been a general disposition among them to publish their methods and experience for the common benefit. For factory processes, a method should be not only reasonably accurate, but also simple and rapid. It is evident, therefore, that the general principles already indicated must underlie any method which would prove useful to factory work. Albert has made a résumé of such methods applicable for factory control, and these are given here for convenience, although they are, in many respects, but condensed statements of methods already described.[98]
[Pg 119] 116. Reagents.—Molybdate Solution.—One hundred and ten grams of pure molybdic acid are dissolved in ammonia of nine-tenths specific gravity and diluted with water to one liter. The solution is poured into one liter of nitric acid, of one and two-tenths specific gravity, and, after standing a few days, filtered.
Concentrated Ammonium Nitrate Solution.—Seven hundred and fifty grams of pure ammonium nitrate are dissolved in water and made up to one liter.
Magnesia Mixture.—Fifty-five grams of magnesium chlorid; seventy grams of ammonium chlorid; 130 cubic centimeters of ammonia of nine-tenths specific gravity are dissolved and diluted with water to one liter.
Two and One-Half Per Cent Ammonia.—One hundred cubic centimeters of ammonia of nine-tenths specific gravity are diluted with water to one liter.
Joulie’s Citrate Solution.—Four hundred grams of citric acid are dissolved in ammonia of nine-tenths specific gravity and diluted to one liter with ammonia of the same strength.
Wagner’s Citrate Solution.—One hundred and fifty grams of citric acid are exactly neutralized with ammonia, then ten grams of citric acid added and diluted to one liter with water.
Sodium Acetate Solution.—One hundred grams of sodium acetate, crystallized, are dissolved in water, treated with 100 cubic centimeters of acetic acid, and diluted to one liter with water.
Calcium Phosphate Solution.—About ten grams of dry, pure tribasic calcium phosphate are dissolved in nitric acid and diluted with water to one liter. In this solution the phosphoric acid is determined gravimetrically by the molybdate or citrate method, and the value of the solution marked on the flask containing it.
Titrated Uranium Solution.—Two hundred and fifty grams of uranium nitrate are dissolved in water, twenty-five grams of sodium acetate added, and the whole diluted to seven liters. One cubic centimeter of this solution corresponds to about 0.005 gram of phosphorus pentoxid. In order to determine its exact value proceed as follows: Twenty-five cubic centimeters of the calcium phosphate solution which, for example, has been found to contain 0.10317 gram of phosphorus pentoxid, are neutralized in a porcelain dish with ammonia, acidified with acetic, treated with ten cubic centimeters of sodium [Pg 120] acetate solution, and warmed. Through a burette as much uranium solution is allowed to flow as is necessary to show in a drop of the solution taken out of the dish, when treated with a drop of pure potassium ferrocyanid, a slight brown color. In order to be certain, this operation is repeated two or three times with new quantities of twenty-five cubic centimeters of calcium phosphate solution. Example:
Twenty-five cubic centimeters of the calcium phosphate solution containing 0.10317 gram of phosphorus pentoxid, gave as a mean of three determinations 23.2 cubic centimeters of the uranium solution necessary to produce the brown color with potassium ferrocyanid. Consequently 0.10317 ÷ 23.2 = 0.00445 gram of phosphorus pentoxid equivalent to one cubic centimeter of uranium solution. If, for instance, a quantity of fertilizer weighing exactly five grams, require ten cubic centimeters of the uranium solution for the complete precipitation of its phosphoric acid, then the quantity of phosphoric acid contained in the fertilizer would be equivalent to 10 × 0.0045, equivalent to 0.0445 gram of phosphorus pentoxid. The fertilizer, therefore, contains eight and nine-tenths per cent of phosphorus pentoxid.
Conduct of the Molybdenum Method.—This method rests upon the precipitation of the phosphorus pentoxid by a solution of ammonium molybdate in nitric acid, solution of the precipitate in ammonia, and subsequent precipitation with magnesia.
Manipulation.—Twenty-five or fifty cubic centimeters of a solution of the phosphate which has been made up to a standard volume and containing about one-tenth gram of phosphorus pentoxid, are placed in a beaker together with 100 cubic centimeters of the molybdate solution and treated with as much ammonium nitrate solution as will be sufficient to give the liquid a content of fifteen per cent of ammonium nitrate. The contents of the beaker are well mixed and warmed for about twenty minutes at from 60° to 80°. After cooling, they are filtered and the precipitate washed on the filter with cold water until a drop of the filtrate saturated with ammonia does not become opaque on treatment with ammonium oxalate. The filtrate is then washed from the filter with two and one-half per cent ammonia solution and precipitated slowly and with constant stirring by the magnesia mixture. After standing for two [Pg 121] hours the ammonium magnesium phosphate is separated by filtration, washed with two and one-half per cent ammonia until the filtrate contains no more chlorin, and ignited.
Conduct of the Citrate Method.—The principle of this method depends upon the fact that when a sufficient quantity of ammonium citrate is added to phosphate solutions, iron, alumina, and lime are retained in solution when, on the addition of the magnesia mixture in the presence of free ammonia, the phosphoric acid is completely precipitated as ammonium magnesium phosphate.
Manipulation.—From ten to fifty cubic centimeters of the solution of the phosphate to be determined are treated with fifteen cubic centimeters of the Joulie citrate solution avoiding warming. A few pieces of filter-paper, the ash content of which is known, are thrown in and, with stirring, fifteen cubic centimeters of magnesia mixture slowly added and if necessary also some free ammonia. By the small pieces of filter-paper the collection of the precipitate against the sides of the vessel and on the stirring rod is prevented and in this way the production of the precipitate hastened. After standing from one-half an hour to two hours the mixture is filtered, ignited, and weighed. If it be preferred to estimate the phosphoric acid by titration, the precipitate is dissolved in a little nitric acid, made slightly alkaline with ammonia, and then acid with acetic and then afterwards titrated with the standard uranium solution.
Conduct of the Uranium Method.—The principle upon which this method rests depends upon the fact that uranium nitrate or acetate precipitates uranium phosphate from solutions containing phosphoric acid and which contain no other free acid except acetic. In the presence of ammonium salts the precipitate is uranium ammonium phosphate having the formula PO₄NH₄UrO₂. The smallest excess of soluble uranium salt is at once detected by the ordinary treatment with potassium ferrocyanid.
Manipulation.—In all cases the solution is first made slightly alkaline with ammonia and then acid by a few drops of acetic, so that no free mineral acid may be present. [Pg 122]
(1) With liquids free from iron:
If, on the addition of ammonium or sodium acetate, no turbidity be produced, the liquid is free from iron and alumina. In this case from ten to fifty cubic centimeters of the solution containing about one-tenth gram of phosphorus pentoxid are treated with ten cubic centimeters of sodium acetate, and afterwards with a quantity of uranium solution corresponding, as nearly as possible, to its supposed content of phosphorus pentoxid, and heated to boiling. From the heated liquid by means of a glass rod, one or two drops are taken and placed upon a porcelain plate and one drop of a freshly prepared solution of potassium ferrocyanid allowed to flow on it. If no brown color be seen at the point of contact of the two drops, additional quantities of the uranium solution are added and, after boiling, again tested with potassium ferrocyanid until a brown color is distinctly visible. The quantity of the uranium solution thus having been determined, duplicate analyses can be made and the whole quantity of the uranium solution added at once with the exception of the last drops, which are added as before.
(2) Solutions containing iron and alumina.
The solution is treated with the ammonium citrate solution of Joulie, the magnesia mixture added slowly, and the precipitate collected on a filter and washed with two and one-half per cent ammonia. The precipitate is then dissolved in nitric acid, made alkaline with ammonia, and then acid with acetic. This solution is then treated with ten cubic centimeters of sodium acetate and titrated with uranium, as described in (1). As an alternative method, 200 cubic centimeters of the superphosphate solution may be treated with fifty cubic centimeters of sodium acetate, allowed to stand for some time, and filtered through a filter of known ash content. In fifty cubic centimeters of the filtrate, which correspond to forty cubic centimeters of the original solution, phosphoric acid may be determined as described above. The precipitate, consisting of iron and aluminum phosphates, is washed three times on the filter with boiling water, dried, and ignited in a platinum dish. The weight of ignited precipitate, diminished by the weight of the ash contained in the filter and divided by two, gives the quantity of phosphorus pentoxid which it is necessary to add to that obtained by titration. [Pg 123]
117. Determination of the Phosphoric Acid in all Phosphates and Basic Slags.—
(1) Total phosphoric Acid:
Five grams of the fine phosphate meal, or slag meal, are moistened in a flask of 500 cubic centimeters content with some water and boiled on a sand-bath with forty cubic centimeters of hydrochloric acid of from 16° to 20° Beaumé. The boiling is continued until only a few cubic centimeters of a thick jelly of silicic acid remain. After cooling, some water is added and the phosphate shaken until the thick lumps of silica are finely divided. The flask is then filled to 500 cubic centimeters and its contents filtered. Fifty cubic centimeters of the filtrate are treated with fifteen cubic centimeters of the Joulie solution and treated in the manner described with magnesia mixture, precipitated, ignited, and weighed. The precipitate can also be dissolved and treated with uranium solution as described.
The method used by Oliveri may also be employed and it is carried out as indicated in the following description:[99]
A weighed quantity of the slag is reduced to a fine powder. To five grams of the sample is added three times its weight of potassium chlorate and the whole is intimately mixed. The mixture is then placed in a porcelain dish and hydrochloric acid is added, little by little, until the potash salt is completely decomposed. It is evaporated until the mass is dry. The material is then treated with fuming nitric acid, and the determination of the phosphorus is made by the ordinary gravimetric method.
By carrying on the operation as described above, a reduction of phosphoric acid is avoided, and the presence of an abundant quantity of potash prevents the formation of basic iron phosphate which is insoluble in nitric acid.
(2) Citrate-Soluble Phosphoric Acid.—One gram of the basic slag or phosphate is placed in a 100 cubic centimeter flask and covered with Wagner’s acid citrate solution making the total volume up to 100 cubic centimeters. With frequent shaking the flask is kept at 40° for an hour, or it may be allowed to stand for twelve hours at room temperature with frequent shaking. In fifty cubic centimeters of the filtrate from this flask the phosphoric acid is determined by the magnesia mixture as described. Since, in the present case, the [Pg 124] precipitate of ammonium magnesium phosphate contains some silicic acid it cannot be directly ignited but must be treated in the following manner: The precipitate and the filter are thrown into a porcelain dish, the filter-paper torn up into shreds with a glass rod, the precipitate dissolved in nitric acid, neutralized with ammonia, acidified with acetic, and treated with uranium solution. The phosphoric acid may also be estimated by the gravimetric method by dissolving the precipitate again in hydrochloric or nitric acid, evaporating to dryness, and drying for one hour at from 110° to 120°, dissolving again in hydrochloric acid, filtering, and washing the precipitate well. The filtrate, which is now free from silica, can be treated with Joulie’s solution, precipitated with magnesia mixture, the precipitate washed, ignited, and weighed as described. The molybdenum method is preferred in the estimation of citrate-soluble phosphoric acid, especially in slags. For this purpose fifty cubic centimeters of the filtrate from the solution of one gram of slag in 100 cubic centimeters of Wagner’s citrate liquid are treated with 100 cubic centimeters of molybdenum solution and thirty cubic centimeters of ammonium nitrate solution, warmed for twenty minutes at 80°, filtered after cooling, and the yellow precipitate washed with cold water. The water will gradually dissolve all the silicic acid from the yellow precipitate and carry it into the filtrate. The yellow precipitate is then dissolved in two and one-half per cent liquid ammonia and precipitated with magnesia mixture and the precipitate washed, ignited, and weighed in the way described.
118. Determination of Phosphoric Acid in Superphosphates.—(1) Citrate-Soluble Phosphoric Acid.—Five grams of the superphosphate are rubbed with 100 cubic centimeters of Wagner’s acid citrate solution in a mortar and washed into a flask of 500 cubic centimeters content and diluted to 500 cubic centimeters with water. With frequent shaking the flask is allowed to stand for twelve hours, after which its contents are filtered. Fifty cubic centimeters of the filtrate are treated with ten cubic centimeters of the Joulie solution and fifteen cubic centimeters of the magnesia mixture and, if necessary, made distinctly alkaline with ammonia, vigorously stirred, [Pg 125] and, after two hours, filtered. The precipitate is washed, ignited, and weighed as described, or titrated, after solution in nitric acid and the addition of sodium acetate, with uranium solution. Example:
The weighed precipitate has 0.1272 gram Mg₂P₂O₇ then the phosphate contains 12.72 × 2 × 0.64 = 16.28 per cent of citrate-soluble P₂O₅.
(2) Water-Soluble Phosphoric Acid.—Twenty grams of superphosphate are rubbed in a mortar and washed into a flask of one liter content and made up to the mark with water. After two hours’ digestion with frequent shaking, the contents of the flask are filtered through a folded filter. Twenty-five cubic centimeters of the filtrate equivalent to five-tenths gram of the substance are precipitated with magnesia mixture, the precipitate filtered, washed, ignited, and weighed, or the moist filtrate may be dissolved upon the filter with a little nitric acid, treated with sodium acetate, and titrated, as described, with uranium solution.
Example: 14.5 cubic centimeters of the uranium solution are required for the precipitate from twenty-five cubic centimeters of the original solution = 0.5 gram superphosphate; it contains then 14.5 × 0.00445 = 0.0645 gram P₂O₅. Consequently the superphosphate contains 12.90 per cent of water-soluble P₂O₅.
Total Phosphoric Add.—Twenty grams of the superphosphate are boiled with fifty cubic centimeters of hydrochloric acid of from 16° to 18° Beaumé for about ten minutes and, after cooling, made up to one liter with water and filtered. Twenty-five cubic centimeters of the filtrate are treated with ten cubic centimeters of Joulie’s citrate solution, a few pieces of filter-paper thrown in, fifteen cubic centimeters of magnesia mixture added, and the whole thoroughly stirred. After standing two hours the contents of the flask are filtered and the precipitate is washed with dilute ammonia and the filter and the precipitate are placed in a platinum crucible. The crucible is heated slowly until the moisture is driven off and the filter burned. Then the temperature is gradually raised to a white heat. The residue is cooled and weighed. Example:
The precipitate weighs, after the subtraction of the filter ash, 0.1390 gram; then the superphosphate contains 13.90 × 2 × 0.64 = 17.79 per cent phosphoric acid. [Pg 126]
119. Time Required for Precipitation of Phosphoric Acid.—The length of time required for the complete precipitation of the phosphoric acid by molybdate mixture is perhaps much less than generally supposed. At 65° the precipitation, as shown by de Roode, is complete in five minutes.[100] In a given case the weight of pyrophosphate obtained after five minutes was 0.0676 gram, and exactly the same weight was found after twenty-four hours. In view of these facts analysts would often be able to save time by omitting the delay usually demanded by the setting aside of the yellow precipitate for a few hours in order to secure a complete separation of the phosphoric acid. In the method of the official chemists it is directed that the digestion at 65° be continued for one hour, and this time may possibly be shortened with advantage. In all cases, however, where there is any doubt in regard to the complete separation, some of the molybdate solution should be added to the filtrate and, with renewed digestion, it should be noted whether any additional precipitate be formed.
120. Examination of the Pyrophosphate.—In fertilizer control it is not usually thought necessary to examine the magnesium pyrophosphate for impurities. Among those most likely to be found is silica. It is proper, in all cases where accuracy is required, to dissolve the precipitate in nitric acid, boil for some time to convert the pyro- into orthophosphate, and reprecipitate with molybdate and magnesia mixture. This treatment will separate the silica which remains practically insoluble after the first ignition. It has been observed by some analysts that the results obtained by the official method are a trifle too high and also that on re-solution the second precipitate of pyrophosphate weighs less than the first.[101] The difference in most cases is very little but it may become a quantity of considerable magnitude in samples where soluble silica is found in notable quantities. The danger of contamination with iron, alumina, and arsenic has already been mentioned but it is not of sufficient importance to warrant further attention. [Pg 127]
121. Iodin in Phosphates.—The presence of iodin has been detected in many natural phosphates and is of interest in the discussion of the problem of their origin.[102] A qualitative test for the detection of iodin may be applied in the following manner: Some finely ground phosphate is mixed with strong sulfuric acid and the gases arising from the reaction are aspired into some carbon disulfid or chloroform. The violet coloration arising indicates the presence of iodin. The gases carrying the iodin may also be brought into contact with starch-paste producing the well-known blue color.
The quantity of iodin present in a phosphate is rarely more than one or two-tenths of one per cent. It can be determined as a silver salt, in the absence of chlorin or by any of the standard methods found in works on qualitative analysis.
Iodin is quite a constant constituent of Florida phosphates.
For a quantitative determination, the sample is treated with an excess of strong sulfuric acid in a closed flask and during the decomposition a stream of air is aspired through the flask and caused to bubble through absorption bulbs containing sodium hydroxid in solution.
The temperature of the decomposition may be raised to about 200°. After the solution of the sample the sodium iodid formed is oxidized by heating with potassium permanganate, acidulated and mixed with a solution of potassium iodid to hold the free iodin in solution. The free iodin is determined in the usual way by titration with standard sodium thiosulfate solution. The reactions preparatory to the titration are represented by the following formulas:
2KI + H₂SO₄ = K₂SO₄ + 2HI.
2HI + H₂SO₄ = 2H₂O + SO₂ + 2I.
6I + 6NaOH = NaIO₃ + 5NaI + 3H₂O.
NaI + 2KMnO₄ + H₂O = NaIO₃ + 2KOH + 2MnO₂.
HIO₃ + 5HI = 6I + 3H₂O.
The titration is represented by the following reaction:
2Na₂S₂O₂ + 2I = 2NaI + 2NaI + Na₂S₄O₄.
The decinormal solution of sodium thiosulfate may be used. Grind the crystals of the salt to a fine powder, dry between blotting papers, and use 24.8 grams of the dried salt per liter. The quantity of iodin found [Pg 128] in phosphates is so minute that it is hardly worth while to make a quantitative determination of it.
122. Occurrence of Chromium in Phosphates.—In some phosphates a small quantity of chromium has been found. In a sample of phosphate from the Island of Los Roques in the Caribbean Sea, Gilbert found three-fourths per cent of chromium oxid (Cr₂O₃). The phosphates containing chromium have a greenish color and are characterized by great insolubility in solutions containing organic acids. The chromium is to be determined by the usual methods described in mineral analysis.
123. Estimation of Vanadium.—In the complete analysis of basic slags it becomes necessary to determine the presence of vanadium and its quantity. The method used in this laboratory for the purpose is the volumetric process of Lindemann.[103] It is conducted as follows: Dissolve four grams of the finely powdered slag in sixty cubic centimeters of dilute sulfuric acid (1 : 4), boil for a few minutes, cool, make the volume up to 100 cubic centimeters, filter, and take an aliquot part for the determination. Add decinormal potassium permanganate solution in slight excess to secure the oxidation of the vanadium to vanadium pentoxid. Add, drop by drop, a weak solution of ferrous sulfate until the pink color just disappears. Prepare a ferrous sulfate solution by dissolving 2.183 grams of piano wire in sulfuric acid and making the volume to one liter. Titrate the vanadic mixture with this solution until a drop of the clear liquor removed and brought in contact with potassium ferricyanid shows a distinctive blue-green color.
One cubic centimeter of the ferrous sulfate solution is equivalent to 0.002 gram of vanadium, 0.002888 gram of vanadium dioxid, and 0.003648 gram of vanadium pentoxid. The ferrous sulfate solution may also be made and standardized by any of the approved methods in common use.
The method described by Blair, designed especially for the estimation of vanadium in iron and steel, is conducted in the following manner:[104] Five grams of the drillings are dissolved in fifty cubic centimeters of nitric acid of 1.24 specific gravity. The solution is evaporated to dryness in a porcelain dish and heated thereafter until the nitrates are nearly decomposed. After cooling, the dried mass is [Pg 129] transferred to a mortar and finely ground with thirty grams of dry sodium carbonate and three grams of sodium nitrate. The finely ground materials are placed in a platinum dish and fused for an hour at a high temperature. Spread the fused mass over the sides of the dish while cooling, and afterwards dissolve in hot water, filter, and wash until the volume is a little over half a liter. Add nitric acid to decompose carbonates, but not completely, and boil to get rid of carbon dioxid, being careful to keep the mass always slightly alkaline. Add nitric acid, drop by drop, until slightly in excess, and then sodium carbonate to marked alkalinity, boil, and filter. Add a slight excess of nitric acid to the filtrate, and the development of a yellow color will indicate the presence of vanadic acid. Add to the solution a small quantity of mercurous nitrate and then an excess of mercuric oxid, suspended in water to render the solution neutral and insure the complete precipitation of mercurous vanadate. The mercurous salt also precipitates phosphoric, chromic, tungstic, and molybdic acids which may be present. Boil, filter, and wash the precipitate with hot water, dry, and ignite. Fuse the residue with sodium carbonate and a little nitrate. Dissolve the fused mass, after cooling, in a little water and filter. Add to the filtrate, ammonium chlorid in excess, from three to five grams for each 100 cubic centimeters of the solution, and allow to stand, with occasional stirring, for some time. Ammonium vanadate, insoluble in a saturated solution of ammonium chlorid, separates as a white powder. It is necessary to keep the solution alkaline, and a drop of ammonia should be added from time to time for this purpose. The appearance of a yellowish tint at any time indicates that the solution has become acid, and this acidity must be corrected, or else the results will be too low. Separate the ammonium vanadate by filtration; wash first with a saturated solution of ammonium chlorid containing a little free ammonia, and then with alcohol. Dry, ignite, and moisten with a few drops of nitric acid; again ignite to obtain the compound as vanadium pentoxid. This compound contains 56.22 per cent of vanadium. The method of Rosenheim and Holversheet may also be used.[105] It is based on the preliminary precipitation of the vanadic acid as a [Pg 130] barium or lead salt. The substance supposed to contain vanadium is first brought into solution in such a manner as to secure it as vanadic acid, which is then precipitated with barium chlorid or lead acetate. The precipitate is boiled with hydrochloric acid and potassium bromid, and the liberated bromin determined by the quantity of iodin set free from potassium iodid. In the absence of bodies, such as molybdic acid, which are reduced by sulfurous acid or hydrogen sulfid, the vanadic acid may also be determined by reducing it with one of these reagents and, after removing the excess by boiling, titrating the vanadium tetroxid with potassium permanganate. When vanadic and phosphoric acids occur together the former may be first reduced to tetroxid with sulfurous acid, and after expelling excess of this reagent, the phosphoric acid may be separated with molybdate solution and removed by filtration. When the amount of vanadic acid is large the phosphoric acid should be separated rapidly at 55°-60°, using a considerable excess of the molybdate; or the vanadic acid may first be determined in the solution volumetrically by the bromin process above described, and afterwards the phosphoric acid obtained by evaporating to dryness with a little sulfuric acid, taking the residue up with water, reducing the vanadic with sulfurous acid and precipitating the phosphoric acid with molybdate solution as described above.
124. Fluorin in Bones.—Fluorin is not only a constituent of mineral phosphates but also of bones. According to the researches of Carnot there is often as much as one-half per cent of calcium fluorid in bones and teeth.[106] Gabriel has suggested a means of determining a minimum limit of fluorin in bones and teeth by the development of etchings in comparison with known quantities of pure calcium fluorid. The minimum quantity of calcium fluorid necessary to produce a distinct etching, in known conditions, having been determined, the test is applied to known weights of ignited bone or teeth. He concludes from his results, that the ash of bones and teeth often contains less than one-tenth per cent of fluorin. Since, however, there is a loss of fluorin from calcium fluorid, on ignition, the whole of the fluorin may not have been available in the tests described. [Pg 131]
125. Note on the Separation of Iron and Aluminum Phosphates from the Calcium Compound.—There are many points of difference noted in the descriptions given by authors of the deportment of the iron, and aluminum phosphates in presence of a large excess of the calcium salt. Especially is this true of the statements made by Hess and Glaser[107] in paragraphs 34 and 35. The subject is of such importance, from an analytical point of view, as to merit a careful study.
In this laboratory a thorough investigation of the mutual deportment of these three phosphates has been made by Brown with the following results:[108] When a mixture containing a known weight of the salts was treated exactly as Hess directs, in no case was there a complete separation of the iron aluminum phosphate from the calcium salt. In order to discover the cause of the failure, pure solutions of calcium and iron aluminum phosphates were treated under identical conditions by the necessary reagents. Fifty cubic centimeters of a solution of calcium phosphate, containing about one gram of the salt, were treated with 100 cubic centimeters of water and fifty cubic centimeters of the commercial ammonium acetate containing 150 grams of the salt in a liter. An immediate precipitate was produced at ordinary temperature, and on heating to 60° it became abundant. The addition of ammonium chlorid, phosphate, and nitrate in successive portions, does not prevent the precipitation. Making the solution more dilute lessens the difficulty when twenty cubic centimeters of a ten per cent solution of ammonium phosphate are first added, followed by the usual quantity of ammonium acetate; a clear crystalline precipitate is sometimes observed. Experience also shows that the trouble is not due to an excess of the ammonium acetate.
In treating a solution of iron aluminum phosphate, in similar circumstances, with the ammonium acetate, it is found that a complete precipitation takes place.
Since diluting the solution of the calcium salt diminishes its tendency to form a precipitate with the ammonium acetate the true method of separation seems to lie in that direction. The calcium salt is held completely in solution when the separation is made in the following way.
The solution containing the mixed phosphates is diluted so as to contain [Pg 132] not more than one gram thereof in half a liter. To this is added one drop of dimethylanilin orange, and afterwards ammonium hydroxid, until a very slight precipitate is formed. The mixture is heated to 70° and from twenty to twenty-five cubic centimeters of a twenty-five per cent solution of acid ammonium acetate are added, enough to change the rose color of the indicator to orange. The iron aluminum phosphate is separated by filtration and washed with a hot five per cent solution of ammonium nitrate.
The washed precipitate shows no impurity due to calcium, as proved by dissolving it, reprecipitating and filtering, adding ammonium hydroxid to the filtrate, and heating for a long time. Sometimes a slight troubling of the clear liquid may be observed which may be due to a slight solubility of the iron aluminum phosphate in washing, an accident that may occur if the temperature be allowed to fall below 70°, but no weighable amount of material is obtained. If due to calcium phosphate, a greater dilution in the first precipitation will remove even this mere trace of that salt. In the above conditions the contamination of the iron aluminum precipitate with calcium phosphate may be entirely avoided. We had also undertaken here the problem of separating the phosphoric acid by the citrate method, followed by a destruction of the citric acid in the filtrate by combustion with sulfuric acid according to the kjeldahl process, and final separation of the iron and alumina in the residues when our attention was called to substantially the same process as described by Jean.[109] The method merits a further critical examination.
126. Phosphoric Acid Soluble in Ammonium Citrate.—There is no other point connected with the determination of phosphoric acid which has excited so much discussion and about which there is such difference of opinion as the solubility of phosphates in ammonium citrate. It was clearly established by Huston, in 1882, that the ammonium citrate, as used in fertilizer analysis, would attack normal tricalcium phosphate as it exists in bones.[110]
In a raw bone, finely ground, containing 20.28 per cent of phosphoric acid, the following quantities were found to be soluble in a neutral ammonium citrate solution of 1.09 specific gravity. [Pg 133]
| Time of digestion, thirty minutes. | ||||
| Temperature | 30° | 40° | 50° | 60° |
| Per cent P₂O₅ dissolved | 2.76 | 4.01 | 3.39 | 5.88 |
From this it appears that the quantity of acid dissolved increases with the temperature of digestion with the exception of the number obtained at 50°. When the time of digestion was increased there was also found a progressive increase in the amount of acid passing into solution. At 40° for forty-five minutes the per cent dissolved was 4.97, and at 40° for one hour it was 5.92. These early determinations had the effect of calling attention to the thoroughly empirical process which was in use, in many modified forms, by agricultural chemists, the world over for determining so-called reverted phosphoric acid in fertilizers. Since the publication of the paper above named many investigations have been undertaken by Huston and others relating to this matter.[111] The general results of these studies, tabulated by Huston, are given below.[112]
Influence of the Time of Digestion.
| Material. | Authority. | (A) | Time of digestion. |
(B) Per cent. |
(C) Per cent. |
||
|---|---|---|---|---|---|---|---|
| Bone meal, | F. B. Dancy, | 65 | ½ | hour | 10.60 | 19.75 | |
| 65 | 1 | “ | 11.28 | 19.75 | |||
| Orchilla guano, | F. B. Dancy, | 65 | ½ | “ | 6.62 | 21.68 | |
| 65 | 1 | “ | 6.85 | ||||
| Navassa rock, | F. B. Dancy | 65 | ½ | “ | 4.64 | 31.27 | |
| 65 | 1 | “ | 4.81 | 31.27 | |||
| Navassa | F. B. Dancy, | 65 | ½ | “ | 9.00 | 11.47 | |
| superphos., | 65 | 1 | “ | 9.21 | 11.47 | ||
| Bone meal, | H. A. Huston, | 40 | ½ | “ | 4.01 | 20.28 | |
| 40 | 1 | “ | 5.92 | 20.28 | |||
| Bone meal, | H. A. Huston and | 65 | ½ | “ | 6.17 | 23.58 | |
| raw, | W. J. Jones, Jr., | 65 | 1 | “ | 6.49 | 23.58 | |
| 65 | 2 | hours | 8.22 | 23.58 | |||
| 65 | 5 | “ | 9.31 | 23.58 | |||
| Steamed bone, | H. A. Huston and | 65 | ½ | hour | 10.59 | 27.67 | |
| W. J. Jones, Jr., | 65 | 1 | “ | 12.21 | |||
| 65 | 2 | hours | 14.61 | ||||
| 65 | 5 | “ | 17.94 | ||||
| 65 | 10 | “ | 19.73 | ||||
| Florida | H. A. Huston and | 65 | ½ | hour | 0.56 | 19.75 | |
| soft rock, | W. J. Jones, Jr., | 65 | 2 | hours | 1.69 | ||
| 65 | 5 | “ | 1.47 | [Pg 134] | |||
| Precipitated | H. A. Huston and | 65 | ¼ | hour | 26.72 | 33.34 | |
| calcium | W. J. Jones, Jr., | 65 | ½ | “ | 27.26 | ||
| phosphate from | 65 | 1 | “ | 27.28 | |||
| glue works | 65 | 2 | hours | 27.29 | |||
| Pamunky | H. A. Huston and | 65 | ½ | hour | 4.43 | 13.84 | |
| phosphate,[113] | W. J. Jones, Jr., | 65 | 1 | “ | 8.28 | ||
| 65 | 2 | hours | 10.34 | ||||
| 65 | 5 | “ | 11.80 | ||||
| 65 | 10 | “ | 12.58 | ||||
| Calcined | H. A. Huston and | 65 | ½ | hour | 21.24 | 45.15 | |
| Redonda, | W. J. Jones, Jr., | 1 | “ | 31.70 | |||
| 2 | hours | 36.92 | |||||
| 5 | “ | 41.00 | |||||
| 10 | “ | 42.70 | |||||
| South | H. A. Huston and | 65 | ½ | hour | 2.82 | 25.51 | |
| Carolina | W. J. Jones, Jr., | 1 | “ | 3.13 | |||
| rock, | 2 | hours | 3.57 | ||||
| 5 | “ | 3.88 | |||||
Influence of Temperature.
| Material. | Authority. | Time of digestion. |
(A) | (B) Per cent. |
(C) Per cent. |
||
|---|---|---|---|---|---|---|---|
| Apatite | T. S. Gladding[114] | ½ | hour | 40 | 0.30 | ||
| Canadian, | ½ | “ | 65 | 0.56 | |||
| Orchilla guano, | T. S. Gladding, | ½ | “ | 40 | 4.63 | ||
| ½ | “ | 65 | 4.81 | ||||
| South Carolina | T. S. Gladding, | ½ | “ | 40 | 1.09 | ||
| river rock, | ½ | “ | 65 | 1.35 | |||
| Navassa rock, | T. S. Gladding, | ½ | “ | 40 | 2.73 | ||
| ½ | “ | 65 | 2.53 | ||||
| Grand | T. S. Gladding, | ½ | “ | 40 | 1.16 | ||
| Connetable, | ½ | “ | 65 | 1.96 | |||
| Redonda, | S. W. Johnson and | ½ | “ | 40 | 1.70 | 36.68 | |
| E. H. Farrington, | ½ | “ | 65 | 1.85 | |||
| South Carolina | S. W. Johnson and | ½ | “ | 40 | 1.32 | 25.48 | |
| rock, | E. H. Farrington, | ½ | “ | 65 | 1.65 | ||
| Orchilla guano, | S. W. Johnson and | ½ | “ | 40 | 4.92 | 21.05 | |
| E. H. Farrington, | ½ | “ | 65 | 5.85 | |||
| Navassa rock, | S. W. Johnson and | ½ | “ | 40 | 4.10 | 29.90 | |
| E. H. Farrington, | ½ | “ | 65 | 4.22 | |||
| Acid Navassa, | S. W. Johnson and | ½ | “ | 40 | 11.95 | 16.50 | |
| E. H. Farrington, | ½ | “ | 65 | 13.53 | [Pg 135] | ||
| Fine-ground | S. W. Johnson and | ½ | “ | 40 | 9.40 | 23.50 | |
| bone, | E. H. Farrington, | ½ | “ | 65 | 12.90 | ||
| South Carolina | C. V. Sheppard, Jr. | ½ | “ | 40 | 1.72 | 24.50 | |
| land rock, | also H. C. White, | ½ | “ | 65 | 2.11 | ||
| Orchilla guano, | C. V. Sheppard, Jr. | ½ | “ | 40 | 6.48 | 15.85 | |
| also H. C. White, | ½ | “ | 65 | 6.75 | |||
| Calcined | C. V. Sheppard, Jr. | ½ | “ | 40 | 5.70 | 44.85 | |
| Redonda, | also H. C. White, | ½ | “ | 65 | 10.20 | ||
| Raw Redonda, | C. V. Sheppard, Jr. | ½ | “ | 40 | 4.49 | 43.79 | |
| also H. C. White, | ½ | “ | 65 | 7.92 | |||
| Acid phosphate, | C. V. Sheppard, Jr. | ½ | “ | 40 | 3.55 | 18.25 | |
| S. C. 10.35 per cent | also H. C. White, | ½ | “ | 65 | 4.05 | ||
| water-soluble, | |||||||
| Acid Navassa, | C. V. Sheppard, Jr. | ½ | “ | 40 | 10.85 | 16.20 | |
| 2.85 per cent | also H. C. White, | ½ | “ | 65 | 11.00 | ||
| water-soluble, | |||||||
| Bone, | H. A. Huston, | ½ | “ | 30 | 2.76 | 20.28 | |
| ½ | “ | 40 | 4.01 | ||||
| ½ | “ | 50 | 3.39 | ||||
| ½ | “ | 60 | 5.88 | ||||
| Acid phosphate, | Sheppard and | ½ | “ | 40 | 3.46 | 15.95 | |
| 11.41 per cent | Robertson, | ½ | “ | 60 | 3.82 | ||
| water-soluble, | |||||||
| Calcined | H. A. Huston, | ½ | “ | 40 | 2.18 | 45.46 | |
| Redonda, | ½ | “ | 50 | 5.52 | |||
| ½ | “ | 65 | 21.24 | ||||
| ½ | “ | 75 | 32.90 | ||||
| ½ | “ | 85 | 39.52 | ||||
| Calcined | H. A. Huston and | 5 | hours | 40 | 26.78 | 42.90 | |
| Redonda, | W. J. Jones, Jr., | 5 | “ | 65 | 38.19 | ||
| 5 | “ | 85 | 41.57 | ||||
| Pamunky | H. A. Huston and | 5 | “ | 40 | 3.10 | 13.84 | |
| phosphate, | W. J. Jones, Jr., | 5 | “ | 65 | 11.80 | ||
| 5 | “ | 85 | 12.82 | ||||
| Raw bone, | H. A. Huston and | 2 | “ | 40 | 5.96 | 23.58 | |
| W. J. Jones, Jr., | 2 | “ | 65 | 8.22 | |||
| 2 | “ | 85 | 8.71 | ||||
| Steamed bone, | H. A. Huston and | 5 | “ | 40 | 16.02 | 27.67 | |
| W. J. Jones, Jr., | 5 | “ | 65 | 20.22 | |||
| 5 | “ | 85 | 20.66 | ||||
| Precipitated | H. A. Huston and | 2 | “ | 40 | 24.14 | 33.34 | |
| calcium phosphate | W. J. Jones, Jr., | 2 | “ | 65 | 23.45 | ||
| from glue works, | 2 | “ | 85 | 22.46 | |||
| Florida | H. A. Huston and | 2 | “ | 40 | 0.00 | 19.75 | |
| soft rock, | W. J. Jones, Jr., | 2 | “ | 65 | 1.69 | ||
| 2 | “ | 85 | 1.99 | [Pg 136] | |||
Influence of Quantity of Material Used.
| Material. | Authority. | Time | (A) | (B) Grams. |
(C) Per cent. |
(D) Per cent. |
||
|---|---|---|---|---|---|---|---|---|
| ⅔ | hour | 40 | 2.0 | 9.94 | 21.68 | |||
| Orchilla guano, | F. B. Dancy, | ⅔ | “ | 40 | 1.0 | 12.14 | ||
| ⅔ | “ | 40 | 0.5 | 13.51 | ||||
| ½ | “ | 65 | 2.0 | 6.62 | ||||
| ½ | “ | 65 | 1.0 | 9.33 | ||||
| Redonda, | S. W. Johnson and | ½ | “ | 40 | 2.0 | 1.70 | 36.68 | |
| E. H. Farrington, | ½ | “ | 40 | 0.4 | 3.46 | |||
| ½ | “ | 65 | 2.0 | 1.85 | ||||
| ½ | “ | 65 | 0.4 | 5.26 | ||||
| South Carolina | S. W. Johnson and | ½ | “ | 40 | 2.0 | 1.32 | 25.48 | |
| rock, | E. H. Farrington, | ½ | “ | 40 | 0.4 | 1.33 | ||
| ½ | “ | 65 | 2.0 | 1.65 | ||||
| ½ | “ | 65 | 0.4 | 3.36 | ||||
| Orchilla guano, | T. S. Gladding, | ½ | “ | 65 | 2.0 | 5.87 | ||
| ½ | “ | 65 | 0.4 | 13.05 | ||||
| Calcined | H. A. Huston, | ½ | “ | 65 | 0.5 | 16.80 | 45.46 | |
| Redonda, | ½ | “ | 65 | 1.0 | 18.26 | |||
| ½ | “ | 65 | 2.0 | 21.24 | ||||
| ½ | “ | 65 | 3.0 | 23.22 | ||||
| ½ | “ | 65 | 5.0 | 24.66 | ||||
| ½ | “ | 65 | 10.0 | 28.64 | ||||
| Calcined | H. A. Huston, | 5 | “ | 65 | 0.5 | 41.77 | 45.46 | |
| Redonda, | 5 | “ | 65 | 2.0 | 41.53 | |||
| 5 | “ | 65 | 8.0 | 39.86 | ||||
| Pamunky | H. A. Huston and | 5 | “ | 65 | 0.5 | 11.81 | 13.84 | |
| phosphate, | W. J. Jones, Jr., | 5 | “ | 65 | 2.0 | 11.80 | ||
| 5 | “ | 65 | 4.0 | 11.44 | ||||
| Raw bone, | H. A. Huston and | 2 | “ | 65 | 0.5 | 16.49 | 23.58 | |
| W. J. Jones, Jr., | 2 | “ | 65 | 2.0 | 8.22 | |||
| 2 | “ | 65 | 4.0 | 7.22 | ||||
| Steamed bone, | H. A. Huston and | 5 | “ | 65 | 0.5 | 26.40 | 27.67 | |
| W. J. Jones, Jr., | 5 | “ | 65 | 2.0 | 17.94 | |||
| 5 | “ | 65 | 4.0 | 12.12 | ||||
| Precipitated | H. A. Huston and | 2 | “ | 65 | 0.5 | 33.34 | 33.34 | |
| calcium phosphate | W. J. Jones, Jr., | 2 | “ | 65 | 2.0 | 27.29 | ||
| from glue works, | 2 | “ | 65 | 4.0 | 19.49 | |||
| Florida | H. A. Huston and | 2 | “ | 65 | 0.5 | 5.50 | 19.75 | |
| soft rock, | W. J. Jones, Jr., | 2 | “ | 65 | 2.0 | 1.69 | ||
| 2 | “ | 65 | 4.0 | 1.27 | [Pg 137] | |||
Influence of Acidity and Alkalinity.
| Material. | Authority. | Time | (A) | (B) | (C) | (D) | (E) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Navassa rock, | T. S. Gladding[115] | ½ | hour | 65 | 0.00 | 0.00 | 2.53 | ||||
| ½ | “ | 65 | 0.733 | gm | 4.87 | ||||||
| ½ | “ | 65 | 0.733 | gm | 1.22 | ||||||
| South Carolina | T. S. Gladding | ½ | “ | 65 | 0.00 | 0.00 | 1.35 | ||||
| rock, | ½ | “ | 65 | 0.733 | “ | 2.89 | |||||
| ½ | “ | 65 | 0.733 | “ | 1.06 | ||||||
| Grand | T. S. Gladding, | ½ | “ | 65 | 0.00 | 0.00 | 1.97 | ||||
| connetable, | ½ | “ | 65 | 0.733 | “ | 1.12 | |||||
| ½ | “ | 65 | 0.733 | “ | 11.44 | ||||||
| Dissolved | H. B. McDonnell, | ½ | “ | 65 | 0.00 | 0.00 | 2.49 | 11.51 | |||
| bone-black and | ½ | “ | 65 | 0.01 | “ | 2.42 | |||||
| cottonseed-meal, | ½ | “ | 65 | 0.01 | “ | 2.37 | |||||
| Ground bone, | H. B. McDonnell, | ½ | “ | 65 | 0.00 | 0.00 | 8.66 | 26.62 | |||
| ½ | “ | 65 | 0.01 | “ | 9.18 | ||||||
| ½ | “ | 65 | 0.01 | “ | 8.00 | ||||||
| Calcined | H. B. McDonnell, | ½ | “ | 65 | 0.00 | 0.00 | 30.61 | 45.11 | |||
| Redonda, | ½ | “ | 65 | 0.01 | “ | 29.42 | |||||
| ½ | “ | 65 | 0.01 | “ | 32.47 | ||||||
| Dissolved | H. A. Huston, | ½ | “ | 65 | 0.00 | 0.00 | 2.24 | 11.32 | |||
| bone-black and | ½ | “ | 65 | 1.00 | “ | 2.24 | |||||
| cottonseed-meal, | ½ | “ | 65 | 1.00 | “ | 2.21 | |||||
| Ground bone, | H. A. Huston and | ½ | “ | 65 | 0.00 | 0.00 | 8.78 | 26.35 | |||
| W. J. Jones, Jr., | ½ | “ | 65 | 1.00 | “ | 13.48 | |||||
| ½ | “ | 65 | 1.00 | “ | 5.35 | ||||||
| Calcined | H. A. Huston and | ½ | “ | 65 | 0.00 | 0.00 | 25.54 | 45.15 | |||
| Redonda, | W. J. Jones, Jr., | ½ | “ | 65 | 1.00 | “ | 18.84 | ||||
| ½ | “ | 65 | 1.00 | “ | 35.20 | ||||||
| South Carolina | H. A. Huston and | ½ | “ | 65 | 0.00 | 0.00 | 1.81 | 27.67 | |||
| rock, | W. J. Jones, Jr., | ½ | “ | 65 | 1.00 | “ | 4.59 | ||||
| ½ | “ | 65 | 1.00 | “ | 0.74 | ||||||
| Basic slag, | H. A. Huston and | ½ | “ | 65 | 0.00 | 0.00 | 6.98 | 19.42 | |||
| W. J. Jones, Jr., | ½ | “ | 65 | 1.0 | “ | 10.12 | |||||
| ½ | “ | 65 | 1.0 | “ | 5.49 | ||||||
| Pamunky | H. A. Huston and | 5 | h’rs | 65 | 0.00 | 0.00 | 11.80 | 13.84 | |||
| phosphate, | W. J. Jones, Jr., | 5 | “ | 65 | 1.0 | “ | 11.79 | ||||
| 5 | “ | 65 | 1.0 | “ | 12.28 | ||||||
| Raw bone, | H. A. Huston and | 2 | “ | 65 | 0.00 | 0.00 | 8.22 | 23.58 | |||
| W. J. Jones, Jr., | 2 | “ | 65 | 1.0 | “ | 11.20 | |||||
| 2 | “ | 65 | 1.0 | “ | 4.02 | [Pg 138] | |||||
| Steamed bone, | H. A. Huston and | 5 | “ | 65 | 0.00 | 0.00 | 17.94 | 27.67 | |||
| W. J. Jones, Jr., | 5 | “ | 65 | 1.0 | “ | 22.55 | |||||
| 5 | “ | 65 | 1.0 | “ | 9.64 | ||||||
| Precipitated | H. A. Huston and | 2 | “ | 65 | 0.00 | 0.00 | 24.20 | 33.34 | |||
| calcium phosphate, | W. J. Jones, Jr., | 2 | “ | 65 | 1.0 | “ | 30.70 | ||||
| from glue works, | 2 | “ | 65 | 1.0 | “ | 20.67 | |||||
| Florida soft | H. A. Huston and | 2 | “ | 65 | 0.00 | 0.00 | 1.69 | 19.75 | |||
| rock, | W. J. Jones, Jr., | 2 | “ | 65 | 1.0 | “ | 3.37 | ||||
| 2 | “ | 65 | 1.0 | “ | 0.72 | ||||||
| Calcined | H. A. Huston and | 5 | “ | 65 | 0.00 | 0.00 | 40.64 | 44.30 | |||
| Redonda, | W. J. Jones, Jr., | 5 | “ | 65 | 1.0 | “ | 40.05 | ||||
| 5 | “ | 65 | 1.0 | “ | 41.01 | ||||||
In the above tabulations no mention is made of the work of Fresenius, Neubauer, and Luck, on whose researches the citrate method is based, but an examination of their original paper shows that the temperature conditions are not carefully enough controlled to justify us in tabulating their results.[116] An attempt has been made to include in the above tables, work made under well-defined conditions, which will illustrate the various points under consideration. While each authority of value upon the subject is represented, no attempt has been made to include all the work done by any of them. One element that seems to have been generally overlooked in discussing the problem is that nearly all results have been obtained from one-half hour’s treatment of the material. This means simply the study of an incomplete reaction, and one which is interrupted while the solution is very rapidly going on. This, of course, is only clearly brought out by a comparison of long-time and short-time work in the various tables. In the opinion of Huston very much more work will have to be done before it can be assumed that we have any very clear knowledge of this subject, and very likely the final result will be that all kinds of goods cannot be examined by the same method. The [Pg 139] fact that half a gram of dicalcium phosphate is instantly soluble in 100 cubic centimeters of citrate solution, at ordinary temperatures, while an equal amount of iron and aluminum phosphate is acted upon very slowly at ordinary temperatures will probably have to be taken into consideration, as well as the fact that dicalcium phosphate is less soluble in hot solutions of ammonium citrate than it is in cold solutions, while the reverse is true of the precipitated iron and aluminum phosphate.
At present, the only conclusion that can be safely drawn from the work, is that it would be unsafe to make any generalization upon the subject until more facts are at hand, except that the present methods are unscientific and, unsatisfactory. As the work progresses, new features present themselves, and in such a way as to show that they must be given careful consideration before drawing any final conclusions in the matter.
127. Arbitrary Determination of Reverted Phosphoric Acid.—The so-called reverted phosphoric acid, that is, the acid insoluble in water and soluble in a solution of ammonium citrate, is the most difficult constituent of commercial fertilizers from the point of view of the scientific analyst. A review of all the standard methods which have been given in the preceding pages for its determination must convince every careful observer that, as a rule, each process is based on arbitrary standards, and can give only concordant results when carried out under strictly unvarying conditions. For this reason there can be no just comparison between the results obtained by different methods, which vary from each other only in slight particulars. When, on the other hand, the processes are radically different, the deviations in data become more pronounced.
In such a condition of affairs the analyst is left to choose between methods. He must be guided in his choice not only by what seems to be the most scientific and accurate process, but also, to a certain extent, by the general practice of his professional brethren. For this country, therefore, it is strongly urged that the methods adopted by the Association of Official Agricultural Chemists, be followed in every detail. [Pg 140]
By the phrase “reverted phosphoric acid,” was originally meant an acid once soluble in water, as CaH₄(PO₄)₂, and afterwards changed to a form insoluble in water, but soluble in ammonium citrate as Ca₂H₂(PO₄)₂. But in practice this has never been the true signification of the term. In the manufacture of acid and superphosphates there is formed, more or less of the dicalcium phosphate, either directly or after a time, and this salt which, in no sense can be called reverted, is entirely soluble in ammonium citrate. The iron and aluminum phosphates are also, to a certain degree, soluble in the same reagent. When an acid phosphate, containing various forms of calcium phosphate, is applied to a soil containing iron and alumina, the soluble parts of the compound tend to become fixed by union with those bases, or by precipitation as Ca₂H₂(PO₄)₂. But it is not alone reverted phosphate formed in this way, which the analyst is called on to determine in a fertilizer, although he may have occasion to treat it in soil analysis.
The expression “reverted phosphoric acid,” therefore, in practice not only includes a dicalcium phosphate, which once may have been the monocalcium salt, but also all of that salt originally existing in the superphosphate, and formed directly during its manufacture, as well as any iron and aluminum phosphates present which are soluble in ammonium citrate. The expression “citrate-soluble” is, therefore, to be preferred to “reverted” phosphoric acid.
In the reversion of the phosphoric acid in superphosphates the iron plays a far more important role than the aluminum sulfate. It was formerly supposed that the reversion took place as indicated in the following formula: 2CaH₄(PO₄)₂ + Fe₂O₃ = 2(CaHPO₄, FePO₄) + 3H₂O, while Wagner affirms that the reverted acid compounds consist of varying quantities of ferric oxid, aluminum oxid, phosphorus pentoxid, and calcium oxid, in various states of combination.[117] The more probable reaction is the following: 3CaH₄(PO₄)₂ + Fe₂(SO₄)₃ + 4H₂O = 2(FePO₄, 2H₃PO₄, 2H₂O) + 3CaSO₄. This reaction can be demonstrated by adding to a superphosphate solution one of a ferric salt. In addition to free phosphoric acid, iron phosphate is separated, which gradually passes into an insoluble form by the abstraction of water due to the crystallization of the gypsum. The alumina present [Pg 141] in a superphosphate seems to have no direct influence on the process of reversion. Its phosphate salt is not acted on by the acid calcium phosphate. Even when a superphosphate solution is treated with alum no precipitation is produced, except on warming, and this disappears when the mass is again cold.
It is therefore not necessary in the process of manufacture to separate the alumina by digestion with a hot soda-lye before treating the mass with sulfuric acid.
In order to avoid the reversion of the phosphoric acid several plans have been proposed. One of the best is to use a little excess of sulfuric acid in the manufacture. This tends to hold the phosphoric acid in soluble form but is objectionable on account of drying, handling, and shipping the fertilizer. During the digestion, moreover, it is important that the temperature does not rise above 120°. Another method consists in adding to the dissolved rock a quantity of common salt chemically equivalent to its iron content. Ammonium sulfate also helps to hold the phosphoric acid water-soluble.
128. Influence of Movement.—The influence of time and temperature of digestion, and of variations in the composition of the ammonium citrate on the quantity of phosphoric acid dissolved by that reagent has been pointed out. Of great importance also in the process is the character of the movement to which the materials are subjected during the digestion. For this reason various mechanical devices have been constructed to secure uniformity of solution. Inasmuch as the temperature factor must also be faithfully observed, the best of these devices are so arranged as to admit of a uniform motion within a bath of water kept at the desired temperature which, by the Association method, is 65°. [Pg 142]

Figure. 8.
Huston’s Digesting Apparatus.
129. Digestion Apparatus for Reverted Phosphates.—The digestion apparatus used by Huston consists of two wheels twenty-five centimeters in diameter, mounted on the same axis, having a clear space of four and one-half centimeters between them.[118] In the periphery of each wheel are cut twelve notches, which are to receive the posts bearing the rings through which the necks of the flasks pass. The posts are held in place by nuts which are screwed down on the faces of the wheel. Should it become necessary to take the apparatus apart, it is only necessary to loosen the nuts and the set screw holding one wheel to the shaft and all the parts can at once be removed. The posts extend ten centimeters beyond the face of the wheels, and the rings are four centimeters in internal diameter. Perforated plates, bearing a cross-bar, and held in place by strong spiral springs attached to the plate and the base of the posts, serve to hold the flasks in place. Each plate has a number stenciled through it for convenience in identifying the flasks when it is time to remove [Pg 143] them. Attached to the outside of each post, close to the outer end, is a heavy wire which passes entirely around the apparatus, serving to keep the plates in place after they are removed from the flasks.
The apparatus is mounted on a substantial framework, thirty-six centimeters high and thirty centimeters wide at the base. The space in which the wheel revolves is fourteen centimeters wide. The base bars connecting the two sides are extended seven centimeters beyond one side, and serve for the attachment of lateral bracing. At the top of the framework, at one side, is attached a heavy bar forty-five centimeters long, which serves to carry the cog gearing which transmits the power. The upright shaft carries a cone pulley to provide for varying the speed. The usual speed is two revolutions a minute for the wheel carrying the flasks. The entire apparatus is made of brass. The details of construction are shown in Fig. 8. Round-bottomed flasks are used, and rubber stoppers are held in place by tying or by a special clamp shown at the lower right-hand of the figure.
When high temperatures are used, the plates and flasks are handled by the hooks shown at the left and right-hand upper corners of the figure.
When any other than room temperature is desired, the whole apparatus is immersed in water contained in the large galvanized tank forming the back-ground of the figures. The tank is seventy-five centimeters long, seventy-five centimeters high, and thirty centimeters wide. At one end, near the top, is an extension to provide space for heating the fluid in the flasks before introducing the solid in such cases as may be desired.
The apparatus is held in place by angle irons soldered to the bottom of the tank and a brace resting against the upright bar bearing the gear-wheels.
The water in the tank is heated by injecting steam, or by burners under the tank. As the tank holds about 300 pounds of water the work is not subject to sudden changes of temperature, and little trouble has been experienced in raising and maintaining the temperature of the water, especially when steam is used.
An electric motor, or a small water-motor with only a very moderate head of water, will furnish ample power. [Pg 144]
130. Comparison of Results.—The following data show the results obtained by the digester as compared with those furnished by the official method, temperature and time of digestion being the same in each instance.
Ammonium Citrate Solution on Phosphates.
| Substance. |
Time of treatment. |
Total phos. acid. Per cent. |
Removed by official method. Per cent. |
Removed by digester. Per cent. |
|
|---|---|---|---|---|---|
| Steamed bone, | ½ | hour | 27.67 | 10.59 | 14.52 |
| 1 | “ | 12.21 | 14.82 | ||
| 2 | hours | 14.61 | 17.56 | ||
| 3½ | “ | 16.48 | 18.53 | ||
| 5 | “ | 17.94 | 20.22 | ||
| 7½ | “ | 18.99 | 20.25 | ||
| 10 | “ | 19.73 | 21.18 | ||
| Marl, | ½ | hour | 13.86 | 4.43 | 4.11 |
| 1 | “ | 8.28 | 6.82 | ||
| 2 | hours | 10.34 | 9.76 | ||
| 3½ | “ | 11.00 | 11.31 | ||
| 5 | “ | 11.80 | 11.83 | ||
| 8 | “ | 12.51 | 12.64 | ||
| 10 | “ | 12.58 | 13.00 | ||
| Acidulated bone, | ½ | hour | 19.38 | 12.09 | 12.28 |
| 1 | “ | 12.47 | 12.40 | ||
| 2 | hours | 12.20 | 12.43 | ||
| 3½ | “ | 12.40 | 12.24 | ||
| 5 | “ | 12.43 | 12.26 | ||
| Bone, | ½ | hour | 21.40 | 6.97 | 8.48 |
| Ammoniated dissolved bone, | ½ | “ | 18.22 | 9.28 | 10.63 |
| Cottonseed-meal and | |||||
| castor pomace, | ½ | “ | 2.52 | 0.23 | 0.25 |
| Phospho bone, | ¼ | “ | 16.55 | 7.06 | 7.21 |
In comparing duplicates, the results from the use of the digester are found to be subject to less variation than those from the usual method.
131. Huston’s Mechanical Stirrer.—The stirring apparatus shown in Fig. 9 differs from those which have heretofore come into use, in requiring but a single belt to drive all the stirring rods, and in having all the parts protected from the laboratory fumes.[119] The details of the belt system are shown in the small diagram in the lower central part of the figure. The apparatus is mounted on a substantial wooden box, 200 centimeters long, thirty centimeters high, and eighteen centimeters wide. The driving pulleys, ten centimeters in diameter, are enclosed in the upper part of the case. The shafts on which these pulleys are mounted extend through the bottom of the enclosing box and carry a wooden disk, eleven centimeters in diameter, to prevent [Pg 145] particles of foreign matter from falling into the beakers. The shafts extend two centimeters below these disks, and to the end of the shafts the bent stirring rods are attached by rubber tubing.
The board forming the support of the driving pulleys is extended two centimeters in front of the apparatus, and in this extension twelve notches are cut, in which are held the corks carrying the tubes which contain the solution to be used in precipitating the material in the beakers.
Figure. 9.
Huston’s Mechanical Stirrer.
The ends of these tubes are drawn out to a fine point so as to deliver the liquid at the rate of about one drop per second.
The front of the apparatus is hinged and permits the whole to be closed when not in use, or during the precipitation.
The apparatus has proven extremely satisfactory in the precipitation of ammonium magnesium phosphate. The precipitate is very crystalline, and where the stirring is continued for some minutes, after the magnesia solution has all been added, no amorphous precipitate is observed on longer standing.
132. The Citrate Method Applied to Samples with Small Content of Phosphoric Acid.—It is well established that the citrate method does not give satisfactory results when applied to samples containing small percentages of phosphoric acid, especially when these are of an organic nature, as for instance, cottonseed cake-meal. In this laboratory attempts have been made to remedy this defect in the process so as to render the use of the method possible even in such cases.[120] Satisfactory results have been obtained by adding to the solution of the cake-meal a definite volume of a phosphate solution of known strength. Solutions of ordinary mineral phosphates are preferred for [Pg 146] this purpose. The following example will show the application of the modified method:
In a sample of cake-meal, (cottonseed cake and castor pomace) the content of phosphoric acid obtained by the molybdate method, was 2.52 per cent.
Determined directly by the citrate method, the following data were obtained:
Allowing to stand thirty hours after adding magnesia mixture, 1.08 and 1.53 per cent in duplicates.
Allowing to stand seventy-two hours after adding magnesia mixture, 2.17 and 2.30 per cent in duplicates.
In each case fifty cubic centimeters of the solution were taken, representing half a gram of the sample.
In another series of determinations twenty-five cubic centimeters of the sample were mixed with an equal volume of a mineral phosphate solution, the value of which had been previously determined by both the molybdic and citrate methods. The fifty cubic centimeters thus obtained represented a quarter of a gram each of the cake-meal and mineral phosphates. The filtration followed eighteen hours after adding the magnesia mixture. The following data show the results of the determinations:
| Per cent P₂O₅ mineral phosphate. |
Per cent P₂O₅ in organic sample. |
Per cent P₂O₅ found in mixture × 2. |
Per cent P₂O₅ in organic sample. |
|
|---|---|---|---|---|
| 1 | 15.37 | 2.52 | 17.90 | 2.53 |
| 2 | 29.16 | 2.52 | 31.68 | 2.52 |
| 3 | 31.37 | 2.52 | 33.83 | 2.45 |
| 4 | 31.58 | 2.52 | 34.20 | 2.62 |
| Mean content of P₂O₅ in organic sample | 2.53 | |||
It is thus demonstrated that the citrate method can be applied with safety even to the determination of the phosphoric acid in organic compounds where the quantity present is less than three per cent. It is further shown that solutions of mineral phosphates varying in content of phosphoric acid from fifteen to thirty-two per cent may be safely used for increasing the content of that acid to the proper degree for complete precipitation. In cases where organic matters are present they should be destroyed by moist combustion with sulfuric acid as in the determination of nitrogen to be described in the next part. [Pg 147]
133. Direct Precipitation of the Citrate-Soluble Phosphoric Acid.—The direct determination of citrate-soluble phosphoric acid by effecting the precipitation by means of magnesia mixture in the solution obtained from the ammonium citrate digestion, has been practiced for many years by numbers of European chemists, and the process has even obtained a place in the official methods of some European countries. Various objections have been urged, however, against the general employment of this method in fertilizer analysis on account of the inaccuracies in the results obtained in certain cases, and it has, therefore, been used to but a very limited extent in this country. Since it is impracticable to effect the precipitation with ammonium molybdate in the presence of citric acid the previous elimination or destruction of this substance has been recognized as essential to the execution of a process involving the separation of the phosphoric acid as phosphomolybdate.
It is evident from the data cited in the preceding paragraph, that great accuracy may be secured in this process by adding a sufficient quantity of a solution of a mineral phosphate and proceeding by the citrate method.
Ross has also proposed to estimate the acid soluble in ammonium citrate directly by first destroying the organic matter by moist combustion with sulfuric acid.[121] He recommends the following process:
After completion of the thirty minutes’ digestion of the sample with citrate solution, twenty-five cubic centimeters are filtered at once into a dry vessel. If the liquid be filtered directly into a dry burette, twenty-five cubic centimeters can be readily transferred to another vessel without dilution. After cooling, run twenty-five cubic centimeters of the solution into a digestion flask of 250-300 cubic centimeters capacity, add about fifteen cubic centimeters of concentrated sulfuric acid and place the flask on a piece of wire gauze over a moderately brisk flame; in about eight minutes the contents of the flask commence to darken and foaming begins, but this will occasion no trouble, if an extremely high, or a very low flame be avoided. In about twelve minutes the foaming ceases and the liquid in the flask [Pg 148] appears quite black; about one grain of mercuric oxid is now added and the digestion is continued over a brisk flame. The operation can be completed in less than half an hour with ease, and in many cases, twenty-five minutes. After cooling, the contents of the flask are washed into a beaker, ammonia is added in slight excess, the solution is acidified with nitric, and after the addition of fifteen grams of ammonium nitrate, the process is conducted as usual.
In case as large an aliquot as fifty cubic centimeters of the original filtrate be used, ten cubic centimeters of sulfuric acid are added, and the digestion is conducted in a flask of 300-500 cubic centimeters capacity; after the liquid has blackened and foaming has progressed to a considerable extent, the flask is removed from the flame, fifteen cubic centimeters more of sulfuric acid are added, and the flask and contents are heated at a moderate temperature for two or three minutes; the mercuric oxid is then added and the operation completed as before described.
Following are some of the advantages offered by the method described:
(1) It dispenses with the necessity of the execution of the frequently tedious operation of bringing upon the filter and washing the residue from the ammonium citrate digestion, while the ignition of this residue together with the subsequent digestion with acid and filtration are also avoided.
(2) It affords a means for the direct estimation of that form of phosphoric acid which, together with the water-soluble, constitutes the available phosphoric acid, thus enabling the latter to be determined by making only two estimations.
(3) In connection with the advantages above mentioned it permits of a considerable saving of time, as well as of labor required in manipulation.
In addition to the tests with mercuric oxid, both potassium nitrate and potassium sulfate were used in the digestion to facilitate oxidation. With the former, several additions of the salt were necessary to secure a satisfactory digestion, and even then the time required was longer than with the mercury or mercuric oxid digestion. With potassium sulfate, the excessive foaming which took place interfered greatly with the execution of the digestion process. [Pg 149]
134. Availability of Phosphatic Fertilizers.—There is perhaps no one question more frequently put to analysts by practical farmers than the one relating to the availability of fertilizing materials. The object of the manufacturer should be to secure each of the valuable ingredients of his goods in the most useful form. The ideal form in which phosphoric acid should come to the soil is one soluble in water. Even in localities where heavy rains may abound, there is not much danger of loss of soluble acid by percolation. As has before been indicated, the soluble acid tends to become fixed in all normal soils, and to remain in a state accessible to the rootlets of plants, and yet free from danger of leaching. For this reason, by most agronomists, the water-soluble acid is not regarded as more available than that portion insoluble in water, yet soluble in ammonium citrate.
In many of the States the statutes, or custom, prescribe that only the water and citrate-soluble acid shall be reckoned as available, the insoluble residue being allowed no place in the estimates of value. In many instances such a custom may lead to considerable error, as in the case of finely ground bones and some forms of soft and easily decomposable tricalcium phosphates. There are also, on the markets, phosphates composed largely of iron and aluminum salts, and these appear to have an available value often in excess of the quantities thereof soluble in ammonium citrate.
As a rule the apatites, when reduced to a fine powder and applied to the soil, are the least available of the natural phosphates. Next in order come the land rock and pebble phosphates which, in most soils, have only a limited availability. The soft fine-ground phosphates, especially in soils rich in humus, have an agricultural value, almost, if not quite equal to a similar amount of acid in the acid phosphates. Fine-ground bones also tend to give up their phosphoric acid with a considerable degree of readiness in most soils. Natural iron and aluminum phosphates, have also, as a rule, a high degree of availability. In each case the analyst must consider all the factors of the case before rendering a decision. Not only the relative solubility of the different components of the offered fertilizer in different menstrua must be taken into consideration, but also the character of [Pg 150] the soil to which it is to be applied, the time of application, and the crop to be grown. By a diligent study of these conditions the analyst may, in the end, reach an accurate judgment of the merits of the sample.
135. Direct Weighing of the Molybdenum Precipitate.—It has already been stated that many attempts have, been made to determine the phosphoric acid by direct weighing as well as by titration, as in the Pemberton method. The point of prime importance in such a direct determination is to secure an ammonium phosphomolybdate mixture of constant composition. Unless this can be done no direct method, either volumetric or gravimetric, can give reliable results. Hanamann[122] proposes to secure this constant composition by varying somewhat the composition of the molybdate mixture and precipitating the phosphoric acid under definite conditions. The molybdate solution employed is prepared as follows:
| Molybdic acid | 100 | grams. |
| Ten per cent ammonia | 1.0 | liter. |
| Nitric acid (1.246 sp. gr.) | 1.5 | liters. |
The precipitation of the phosphoric acid is conducted in the cold with constant stirring. It is complete in half an hour. The ammonium phosphomolybdate is washed with a solution of ammonium nitrate and then with dilute nitric acid, dried, and ignited at less than a red heat. It should then have a bluish-black color throughout. Such a body contains 4.018 per cent of phosphoric anhydrid.
Twenty-five cubic centimeters of a sodium phosphate solution containing fifty milligrams of phosphoric acid, treated as above, gave a bluish-black precipitate weighing 1.249 grams, which, multiplied by 0.041018, equaled 50.018 milligrams of phosphorus pentoxid. The method should be tried on phosphates of various kinds and contents of phosphorus pentoxid before a definite judgment of its merits is formed.
136. Reactions with Phosphates.—In this country the expressions “acid” and “super” phosphates are used interchangeably. A more correct use of the terms would designate by “acid” the phosphate formed directly from tricalcium phosphate by the action of sulfuric acid, [Pg 151] while by “super” would be indicated a similar product formed by the action of free phosphoric acid on the same materials. In Germany the latter compound is called double phosphate.
The reaction which takes place in the first instance is represented by the following formula:
3Ca₃(PO₄)₂ + 6H₂SO₄ + 12H₂O = 4H₃PO₄ + Ca₃(PO₄)₂ + 6(CaSO₄·2H₂O);
and 4H₃PO₄ + Ca₃(PO₄)₂ + 3H₂O = 3[CaH₄(PO₄)₂·H₂O].
A simpler form of the reaction is expressed as follows:
Ca₃(PO₄)₂ + 2H₂SO₄ + 5H₂O = CaH₄(PO₄)₂·H₂O + 2[CaSO₄·2H₂O].
If 310 parts, by weight, of fine-ground tricalcium phosphate be mixed with 196 parts of sulfuric acid and ninety parts of water, and the resulting jelly be quickly diluted with a large quantity of water, and filtered, there will be found in the filtrate about three-quarters of the total phosphoric as free acid. If, however, the jelly, at first, formed as above, be left to become dry and hard, the filtrate, when the mass is beaten up with water and filtered, will contain monocalcium phosphate, CaH₄(PO₄)₂.
If the quantity of sulfuric acid used be not sufficient for complete decomposition, the dicalcium salt is formed directly according to the following reaction:
Ca₃(PO₄)₂ + H₂SO₄ + 6H₂O = Ca₂H₂(PO₄)₂·4H₂O + CaSO₄·2H₂O.
This arises, doubtless, by the formation, at first, of the regular monocalcium salt and the further reaction of this with the tricalcium compound, as follows:
CaH₄(PO₄)₂ + H₂O + Ca₃(PO₄)₂ + 7H₂O = 2[Ca₂H₂(PO₄)₂·4H₂O].
This reaction represents, theoretically, the so-called reversion of the phosphoric acid. When there is an excess of sulfuric acid there is a complete decomposition of the calcium salts with the production of free phosphoric acid and gypsum. The reaction is represented by the following formula:
Ca₃(PO₄)₂ + 3H₂SO₄ + 6H₂O = 2H₃PO₄ + 3[CaSO₄·2H₂O].
[Pg 152] The crystallized gypsum absorbs the six molecules of water in its molecular structure.
137. Reactions with Fluorids.—Since calcium fluorid is present in nearly all mineral phosphates, the reactions of this compound must be taken into consideration in a chemical study of the manufacture of acid phosphates. When treated with sulfuric acid the first reaction which takes place consists in the formation of hydrofluoric acid: CaF₂ + H₂SO₄ = 2HF + CaSO₄. Since, however, there is generally some silica in reach of the nascent acid, all, or a portion of it, combines at once with this silica, forming silicon tetrafluorid: 4HF + SiO₂ = 2H₂O + SiF₄. This compound, however, is decomposed at once in the presence of water, forming hydrofluosilicic acid: 3SiF₄ + 2H₂O = SiO₂ + 2H₂SiF₆. The presence of calcium fluorid in natural phosphates is extremely objectionable from a technical point of view, both on account of the increased consumption of oil of vitriol which it causes, but also by reason of the injurious nature of gaseous fluorin compounds produced. Each 100 pounds of calcium fluorid entails the consumption of 125.6 pounds of sulfuric acid.
138. Reaction with Carbonates.—Most mineral phosphates contain calcium carbonate in varying quantities. This compound is decomposed on treatment with sulfuric acid according to the reaction: CaCO₃ + H₂SO₄ = CaSO₄ + H₂O + CO₂. When present in moderate amounts, calcium carbonate is not an objectionable impurity in natural phosphates intended for acid phosphate manufacture. The reaction with sulfuric acid which takes place produces a proper rise in temperature throughout the mass, while the escaping carbon dioxid permeates and lightens the whole mass, assisting thus in completing the chemical reaction by leaving the residual mass porous, and capable of being easily dried and pulverized. Where large quantities of carbonate in proportion to the phosphate are present the sulfuric acid used should be dilute enough to furnish the necessary water of crystallization to the gypsum formed. For each 100 parts, by weight, of calcium carbonate, eighty parts of sulfuric anhydrid are necessary, or 125 parts of acid of 1.710 specific gravity = 60° Beaumé.
In some guanos a part of the calcium is found as pyrophosphate, and this is acted upon by the sulfuric acid in the following way: Ca₂P₂O₇ + H₂SO₄ = CaH₂P₂O₇ + CaSO₄.
[Pg 153] 139. Solution of the Iron and Alumina Compounds.—Iron may occur in natural phosphates in many forms. It probably is most frequently met with as ferric or ferrous phosphate, seldom as ferric oxid, and often as pyrite, FeS₂. The iron also may sometimes exist as a silicate. The alumina is found chiefly in combination with phosphoric acid, and as silicate.
Where a little less sulfuric acid is employed, as is generally the case, than is necessary for complete solution, the iron phosphate is attacked as represented below:
3FePO₄ + 3H₂SO₄ = FePO₄·2H₂PO₄ + Fe₂(SO₄)₃.
When an excess of sulfuric acid is employed, the formula is reduced to the simple one:
2FePO₄ + 3H₂SO₄ = 2H₃PO₄ + Fe₂(SO₄)₃.
A part of the iron sulfate formed reacts with the acid calcium phosphate present to produce a permanent jelly-like compound, difficult to dry and handle. As much as two per cent of iron phosphate, however, may be present without serious interference with the commercial handling of the product. By using more sulfuric acid as much as four or five per cent of the iron phosphate can be held in solution. Larger quantities are very troublesome from a commercial point of view. The reaction of the ferric sulfate with monocalcium phosphate, is as follows:
3CaH(PO₄)₂ + Fe₂(SO₄)₃ + 4H₂O = 2(FePO₄·2H₃PO₄·2H₂O) + 3CaSO₄.
Pyrite and the silicates containing iron are not attacked by sulfuric acid, and these compounds are therefore left, in the final product, in a harmless state. If the pyritic iron is to be brought into solution aqua regia should be employed.
With sufficient acid the aluminum phosphate is decomposed with the formation of aluminum sulfate and free phosphoric acid:
AlPO₄ + 3H₂SO₄ = Al₂(SO₄)₃ + 2H₃PO₄.
140. Reaction with Magnesium Compounds.—The mineral phosphates, as a rule, contain but little magnesia. When present it is probably as an acid salt, MgHPO₄. Its decomposition takes place in slight deficiency or excess of sulfuric acid respectively as follows:
2MgHP₄ + H₂SO₄ + 2H₂O = [MgH₄(PO₄)₂·2H₂O] + MgSO₄
and MgHPO₄ + H₂SO₄ = H₃PO₄ + MgSO₄.
The magnesia, when in the form of oxid, is capable of producing a [Pg 154] reversion of the monocalcium phosphate, as is shown below:
CaH₄(PO₄)₂ + MgO = CaMgH₂(PO₄)₂ + H₂O.
One part by weight of magnesia can render three and one-half parts of soluble monocalcium phosphate insoluble.
141. Determination of Quantity of Sulfuric Acid Necessary for Solution of a Mineral Phosphate.—The theoretical quantity of sulfuric acid required for the proper treatment of any phosphate may be calculated from its chemical analysis and by the formulas and reactions already given. For the experimental determination the method of Rümpler may be followed.[123]
Twenty grams of the fine phosphate are placed in a liter flask with a greater quantity of accurately measured sulfuric acid than is necessary for complete solution. The acid should have a specific gravity of 1.455 or 45° B. The mixture is allowed to stand for two hours at 50°. It is then cooled, the flask filled with water to the mark, well shaken, and the contents filtered. Fifty cubic centimeters of the filtrate are treated with tenth normal soda-lye until basic phosphate begins to separate. The excess of acid used is then calculated. Example: Twenty grams of phosphate containing 28.3 per cent of phosphoric acid, 10.0 per cent of calcium carbonate, 5.5 per cent of calcium fluorid, and 2.4 per cent of calcium chlorid were treated as above with sixteen cubic centimeters of sulfuric acid containing 10.24 grams of sulfur trioxid. In titrating fifty cubic centimeters of the filtrate obtained as described above, 10.4 cubic centimeters of tenth normal soda-lye were used, equivalent to 0.0416 gram of sulfur trioxid. Then 10.24 × 50 ÷ 1000 = 0.5120 = total sulfur trioxid in fifty cubic centimeters of the filtrate, and 0.5120 - 0.0416 = 0.4704 gram, the amount of sulfur trioxid consumed in the decomposition.
Therefore the sulfur trioxid required for decomposition is 47.04 per cent of the weight of the phosphate employed. One hundred parts of the phosphate would therefore require 47.04 parts of sulfur trioxid = to 73.6 parts of sulfuric acid of 1.710 specific gravity or 92.1 parts of 1.530 specific gravity. [Pg 155]
A more convenient method than the one mentioned above consists in treating a small quantity of the phosphate, from one-half to one kilogram, in the laboratory, or fifty kilograms in a lead box, just as would be practiced on a large scale. A few tests with these small quantities, followed by drying and grinding will reveal to the skilled operator the approximate quantity and strength of sulfuric acid to be used in each case. The quantities of sulfuric acid as determined by calculation from analyses and by actual laboratory tests agree fairly well in most instances. There is, however, sometimes a marked disagreement. The general rule of practice is to use always an amount of sulfuric acid sufficient to produce and maintain water-soluble phosphoric acid in the fertilizer, but the sulfuric acid must not be used in such quantity as to interfere with the subsequent drying, grinding, and marketing of the acid phosphate.
For convenience the following table may be used for calculating the quantity of oil of vitriol needed for each unit of weight of material noted:
One Part by Weight of Each Substance Below Requires:
| Sulfuric Acid by Same Unit of Weight. | |||||
|---|---|---|---|---|---|
| At 48° B. | At 50° B. | At 52° B. | At 54° B. | At 55° B. | |
| Tricalcium phosphate | 1.590 | 1.517 | 1.446 | 1.382 | 1.352 |
| Iron phosphate | 1.630 | 1.558 | 1.485 | 1.420 | 1.390 |
| Aluminum phosphate | 2.025 | 1.930 | 1.839 | 1.756 | 1.721 |
| Calcium carbonate | 1.640 | 1.565 | 1.495 | 1.428 | 1.411 |
| Calcium fluorid | 2.006 | 2.010 | 1.916 | 1.830 | 1.794 |
| Magnesium carbonate | 1.940 | 1.860 | 1.775 | 1.690 | 1.660 |
Example.—Suppose for example a phosphate of the following composition is to be treated with sulfuric acid; viz.,[124]
| Moisture and organic | 4.00 | per cent. |
| Calcium phosphate | 55.00 | “ |
| Calcium carbonate | 3.00 | “ |
| Iron and aluminum phosphate | ||
| nearly all alumina | 6.50 | “ |
| Magnesium carbonate | 0.75 | “ |
| Calcium fluorid | 2.25 | “ |
| Insoluble | 28.00 | “ |
Using sulfuric acid of 50° B., the following quantities will be required for each 100 kilograms. [Pg 156]
| Kilos of acid required. |
||
| Calcium | phosphate, fifty-five kilos | 83.44 |
| “ | carbonate three and a half kilos | 5.48 |
| “ | fluorid, two and a quarter “ | 4.52 |
| Aluminum and iron phosphate, six and a half kilos | 12.55 | |
| Magnesium carbonate, three-quarters of a kilo | 1.40 | |
| Total | 107.39 | |
142. Phosphoric Acid Superphosphates.—If a mineral phosphate be decomposed by free phosphoric in place of sulfuric acid the resulting compound will contain about three times as much available phosphoric acid as is found in the ordinary acid phosphate. The reaction takes place according to the following formulas:
(1) Ca₃(PO₄)₂ + 4H₃PO₄ + 3H₂O = 3[CAH₄(PO₄)₂·H₂O].
(2) Ca₃(PO₄)₃ + 2H₃PO₄ + 12H₂O = 3[Ca₂H₂(PO₄)₂·4H₂O].
In each case the water in the final product is probably united as crystal water with the calcium salts produced. The monocalcium salt formed in the first reaction is soluble in water and the dicalcium salt in the second reaction in ammonium citrate. Where fertilizers are to be transported to great distances there is a considerable saving of freight by the use of such a high-grade phosphate, which may, at times, contain over forty per cent of available acid. The phosphoric acid used is made directly from the mineral phosphate by treating it with an excess of sulfuric acid.
[1] Day, Mineral Resources of the United States 193, pp. 703, et seq.
[2] Massachusetts Agricultural Experiment Station, Bulletin 51, March, 1894.
[3] Brown, Manual of Assaying, p. 24.
[4] Bulletin de l’Association des Chimistes de Sucrèrie, No. 2, pp. 7, et seq.
[5] Proceedings of the Twelfth and Thirteenth Meetings of the Society for the Promotion of Agricultural Science, p. 140.
[6] Chemical Division, U. S. Department of Agriculture, Bulletin 43, p. 341.
[7] Rapport adressé par le Comité des Stations agronomiques au sujet des Methodes à suivre dans l’Analyse des Matières fertilisantes.
[8] Die Landwirtschaftlichen Versuchs-Stationen, Band 38, S. 303.
[Pg 157]
[9] Vid. op. cit. 6, p. 341.
[10] Zeitschrift für analytische Chemie, 1890, S. 390.
[11] Vid. op. cit. 6, p. 342.
[12] Chemisches Centralblatt, Band 2, S. 813.
[13] Transactions of the American Institute of Mining Engineers, Vol. 21, p. 165.
[14] Phosphates of America, p. 144.
[15] Vid. op. et loc. cit. 13.
[16] U. S. Geological Survey, Bulletin No. 47.
[17] Vid. op. et loc. cit. 14.
[18] Vid. op. et loc. cit. 13.
[19] Vid. op. cit. 14, p. 147.
[20] Transactions of the American Institute of Mining Engineers, Vol. 21, p. 168.
[21] Phosphates of America, p. 153.
[22] Die Landwirtschaftlichen Versuchs-Stationen, Band 34, S. 379.
[23] Zeitschrift für analytische Chemie, 1892, S. 383.
[24] Zeitschrift für angewandte Chemie, 1894, Ss. 679 und 701.
[25] Vid. op. cit. supra, 1889, p. 636.
[26] Vid. op. cit. 24, 1891, p. 3.
[27] Rapports presentèes au Congrès International de Chimie Appliqué, Bruxelles, Août, 1894, p. 26.
[28] Vid. op. et loc. cit. 20.
[29] Le Stazioni Sperimentali Agrarie Italiane, Vol. 23, p. 31.
[30] Crookes’ Select Methods, p. 538.
[31] Journal of Analytical and Applied Chemistry, Vol. 5, p. 671. For additional authorities on these methods consult Meyer and Wohlrab, Zeitschrift für angewandte Chemie, 1891, Ss. 170 und 243. Gruber, Zeitschrift für analytische Chemie, Band 30, S. 206. Shephard, Chemical News, May 29, 1891, p. 251. Vögel, Zeitschrift für angewandte Chemie, 1891, Band 12, S. 357.
[32] Journal of the American Chemical Society, April, 1895.
[33] Vid. op. cit. 21, p. 150.
[34] Transactions of the American Institute of Mining Engineers, Vol. 21, p. 170.
[35] Vid. op. cit. supra, p. 173.
[36] Comptes rendus, Tome 54, p. 468.
[37] Crookes’ Select Methods, p. 500.
[38] For details of method see Fresenius quantitative Analysis.
[39] U. S. Department of Agriculture, Chemical Division, Bulletin 43, p. 341.
[40] Letter to B. W. Kilgore, Reporter for Phosphoric Acid to the Association of Official Agricultural Chemists.
[41] Die Landwirtschaftlichen Versuchs-Stationen, Band 38, S. 304.
[42] Communicated by Dr. Solberg.
[Pg 158]
[43] From the Official Swedish Methods; translated for the author by F. W. Woll.
[44] Methoden van Onderzock aan de Rijkslandbouw-proefstations, 1893, p. 4.
[45] Zeitschrift für analytische Chemie, 1893, S. 64.
[46] Journal of the American Chemical Society, Vol. 16.
[47] Zeitschrift für angewandte Chemie, 1894, S. 678.
[48] Journal für Landwirtschaft, Band 30, S. 23.
[49] Vid. op. cit. 47, p. 544.
[50] Vid. op. cit. 46, Vol. 16, p. 462.
[51] Vid. op. et loc. cit. supra.
[52] Die Agricultur-Chemische Versuchs-Station, Halle a/S., Ss. 56, et seq.
[53] Chemische Industrie, 1890.
[54] Vid. op. et loc. cit. 52.
[55] Chemiker Zeitung, 1890, No. 75, S. 1246.
[56] Vid. op. cit. 43.
[57] Glaser, Zeitschrift für analytische Chemie, 24, 178 (1885). Laubheimer, Ibid, 25, 416 (1886). Müller, Tagebl. d. Naturforscher-Vers. zu Wiesbaden, 1886, 365. Vögel, Chemiker Zeitung, 1888, 85. Stutzer, Ibid, 492. Seifert, Ibid, 1390. v. Reis, Zeitschrift für angewandte Chemie, 1888, 354. Loges, Reportorium für analytische Chemie, 7, 85 (1887). Kassuer, Zeitschrift für Nahrungsmitteluntersuchung und Hygiene, 2, 22 (1888). C. Müller, Die Landwirtschaftlichen Versuchs-Stationen, 35, 438 (1888).
[58] L’Engrais, Tome 9, p. 928.
[59] Vid. op. et loc. cit. 44.
[60] Journal of the American Chemical Society, Vol. 16, p. 462.
[61] Die Landwirtschaftlichen Versuchs-Stationen, Band 41, S. 329.
[62] Journal of Analytical and Applied Chemistry, Vol. 5, p. 685.
[63] Vid. op. cit. supra, Vol. 3, p. 413.
[64] Zeitschrift für angewandte Chemie, 1886, S. 354.
[65] Vid. op. cit. 52, p. 61.
[66] Vid. op. cit. 55, Vol. 18, p. 1153.
[67] Chemiker Zeitung, 1894, No. 88, p. 1934.
[68] Op. cit. supra, 1892, p. 1471.
[69] Vid. op. cit. 60, p. 721.
[70] Zeitschrift für analytische Chemie, Band 29, S. 408.
[71] Mitteilungen der deutschen Landwirtschafts Gesellschaft, 1890-’91, No. 11, S. 131.
[72] Zeitschrift für angewandte Chemie, 1888, S. 299.
[73] Vid. op. cit. 70, p. 409.
[74] Vid. op. cit. 72, 1890, p. 595.
[75] Vid. op. cit. 61, Tome 43, p. 183.
[76] Chemiker Zeitung, Band 18, S. 565.
[Pg 159]
[77] Chemical News, Vol. 1, p. 97.
[78] Archive für Wissenschaftliche Heilkunde, Band 4, S. 228.
[79] Journal für praktische Chemie, Band 70, S. 104.
[80] Sutton’s Volumetric Analysis, p. 237.
[81] Bulletin de la Société des Agriculteurs de France, 1876, p. 53.
[82] Manual Agenda des Fabricants de Sucre, 1889, p. 307.
[83] Journal of the American Chemical Society, Vol. 15, p. 382, and Vol. 16, p. 278.
[84] Chemical News, Vol. 47, p. 127.
[85] American Chemical Journal, Vol. 11, p. 84.
[86] Vid. op. cit. 83, Vol. 16, p. 282.
[87] Bulletin 43, Chemical Division, U. S. Department of Agriculture, p. 88.
[88] Vid. op. cit. supra, p. 91.
[89] Repertoire de Pharmacie, 1893, p. 153.
[90] Revue de Chimie Analytique Appliqué, 1893, p. 113.
[91] Chemiker Zeitung, 1894, S. 1533.
[92] Blair, Analysis of Iron and Steel, p. 95.
[93] Journal of Analytical and Applied Chemistry, Vol. 7, p. 108.
[94] Journal of the American Chemical Society, Vol. 17, p. 129.
[95] Vid. op. cit. 92, p. 99.
[96] Eighth Annual Report of Purdue University, p. 238.
[97] Receuil des Travaux Chimiques, Tome 12, pp. 1, et seq. Journal of the Chemical Society (Abstracts), Vol. 64, p. 496.
[98] Zeitschrift für angewandte Chemie, 1891, Ss. 279, et seq.
[99] Le Stazioni Sperimentali Agrarie Italiane, February, 1891.
[100] Journal of the American Chemical Society, Vol. 17, p. 43.
[101] Vid. op. et loc. cit. supra.
[102] L’Engrais, Tome 10, p. 65.
[103] Journal of Analytical and Applied Chemistry, Vol. 5, p. 694. Zeitschrift für analytische Chemie, Band 18, S. 99.
[104] Vid. op. cit. 92, p. 103.
[105] Journal of the Chemical Society (Abstracts), Vol. 58, p. 1343.
[106] Comptes rendus, Tome 114, p. 1189.
[107] Vid. op. cit. 24 and 25.
[108] Report communicated to author by W. G. Brown.
[109] Chemisches Centralblatt, 1895, p. 562.
[110] Wiley, Report on Fertilizers to Indiana State Board of Agriculture, 1882.
[Pg 160]
[111] Proceedings of the Association of Official Agricultural Chemists, Atlanta, 1884, p. 19. Report of Indiana State Board of Agriculture, 1882, p. 230, and Proceedings of the Association of Official Agricultural Chemists, Atlanta, 1884, p. 30. Huston and Jones. (These gentlemen are now investigating all materials used as sources of phosphoric acid in fertilizers; their results here quoted are from unpublished work, and include but a small part of the work so far done.) American Chemical Journal, March, 1884, p. 1. Proceedings of the Association of Official Agricultural Chemists, Atlanta, 1884, p. 23. Ibid, p. 28. Ibid, p. 38. Ibid, p. 45. U. S. Department of Agriculture, Chemical Division, Bulletin No. 7, p. 18. Ibid, Bulletin No. 28, p. 171. Ibid, Bulletin No. 31, p. 100. Ibid, Bulletin No. 31, p. 99.
[112] Manuscript communication to author.
[113] Pamunky phosphate is the so-called “olive earth” found along the Pamunky river, in Virginia. It is almost all precipitated iron and aluminum phosphates, and the product is peculiar in that the iron is almost all in the ferrous condition.
[114] In the work of T. S. Gladding only fifty cubic centimeters of citrate were used.
[115] In the work of T. S. Gladding only fifty cubic centimeters of citrate were used.
[116] Zeitschrift für analytische Chemie, Band 10, S. 133.
[117] Lehrbuch der Düngerfabrication.
[118] Bulletin 54, Purdue Agricultural Experiment Station, p. 4.
[119] Vid. op. cit. supra, p. 7.
[120] Runyan and Wiley; Paper presented to Washington Section of the American Chemical Society, April 11, 1895.
[121] Bulletin 38, Chemical Division, U. S. Department of Agriculture, p. 16.
[122] Chemiker Zeitung, 1895, S. 553.
[123] Die Käuflichen Dungermittel Stoffe, dritte Auflage, 1889.
[124] Wyatt, Phosphates of America, p. 128.
[Pg 161]
143. Kinds of Nitrogen in Fertilizers.—Nitrogen is the most costly of the essential plant foods. It has been shown in the first volume, paragraph 23, that the popular notion regarding the relatively great abundance of nitrogen is erroneous. It forms only 0.02 per cent of the matter forming and pertaining to the earth’s crust. The great mass of nitrogen forming the bulk of the atmosphere is inert and useless in respect of its adaptation to plant food. It is not until it becomes oxidized by combustion, electrical discharges, or the action of certain microorganisms that it assumes an agricultural value.
Having already, in the first volume, described the relation of nitrogen to the soil it remains the sole province of the present part to study it as aggregated in a form suited to plant fertilization. In this function nitrogen may claim the attention of the analyst in the following forms:
1. In organic combination in animal or vegetable substances, forming a large class of bodies, of which protein may be taken as the type. Dried blood or cottonseed-meal illustrates this form of combination.
2. In the form of ammonia or combinations thereof, especially as ammonium sulfate, or as amid nitrogen.
3. In a more highly oxidized form as nitrous or nitric acid usually united with a base of which Chile saltpeter may be taken as a type.
The analyst has often to deal with single forms of nitrogenous compounds, but in many instances may also find all the typical forms in a single sample. Among the possible cases which may arise the following are types:
a. The sample under examination may contain nitrogen in all three forms mentioned above. [Pg 162]
b. There may be present nitrogen in the organic form mixed with nitric nitrogen.
c. Ammoniacal nitrogen may replace the nitric in the above combination.
d. The sample may contain no organic but only nitric and ammoniacal nitrogen.
e. Only nitric or ammoniacal nitrogen may be present.
144. Determination of the State of Combination.—Some of the sample is mixed with a little powdered soda-lime. If ammoniacal nitrogen be present free ammonia is evolved even in the cold and may be detected either by its odor or by testing the escaping gas with litmus or turmeric paper. A glass rod moistened with strong hydrochloric acid will produce white fumes of ammonium chlorid when brought near the escaping ammonia.
If the sample contain any notable amount of nitric acid it will be revealed by treating an aqueous solution of it with a crystal of ferrous sulfate and strong sulfuric acid. The iron salt should be placed in a test-tube with a few drops of the solution of the fertilizer and the sulfuric acid poured down the sides of the tube in such a way as not to mix with the other liquids. The tube must be kept cold. A dark brown ring will mark the disk of separation between the sulfuric acid and the aqueous solution in case nitric acid be present. If water produce a solution of the sample too highly colored to be used as above, alcohol of eighty per cent strength may be substituted. The coloration produced in this case is of a rose or purple tint.
Nitric nitrogen may also be detected by means of brucin. If a few drops of an aqueous solution of brucin be mixed with the same quantity of an aqueous extract of the sample under examination and strong sulfuric acid be added, as described above, there will be developed at the disk of contact between the acid and the mixed solutions a persistent rose tint varying to yellow.
To detect the presence of organic albuminoid nitrogen the residue insoluble in water, when heated with soda-lime, will give rise to ammonia which may be detected as described above. [Pg 163]
145. Microscopic Examination.—If the chemical test reveal the presence of organic nitrogen the next point to be determined is the nature of the substance containing it. Often this is revealed by simple inspection, as in the case of cottonseed-meal. Frequently, however, especially in cases of fine-ground mixed goods, the microscope must be employed to determine the character of the organic matter. It is important to know whether hair, horn, hoof, and other less valuable forms of nitrogenous compounds have been substituted for dried blood, tankage, and more valuable forms. In most cases the qualitative chemical, and microscopic examination will be sufficient. There may be cases, however, where the analyst will be under the necessity of using other means of identification suggested by his skill and experience or the circumstances connected with any particular instance. In such cases the general appearance, odor, and consistence of the sample may afford valuable indications which will aid in discovering the origin of the nitrogenous materials.
146. Seeds and Seed Residues.—The proteid matters in seeds and seed residues, after the extraction of the oil, are highly prized as sources of nitrogenous fertilizers either for direct application or for mixing. Typical of this class of substances is cottonseed-meal, the residue left after the extraction of the oil which is accomplished at the present time mostly by hydraulic pressure. The residual cakes contain still some oil but nearly half their weight consists of nitrogenous compounds. The following table gives the composition of a sample of cottonseed-meal:
| Ash | 7.60 | per cent. |
| Fiber | 4.90 | “ |
| Oil | 10.01 | “ |
| Protein | 51.12 | “ |
| Digestible carbohydrates, etc. | 26.37 | “ |
While the above shows the composition of a single sample of the meal it should be remembered that there may be wide variations from this standard due either to natural composition or to different degrees of the extraction of the oil. [Pg 164]
The composition of the ash is given below:
| Phosphoric acid, | P₂O₅ | 31.01 | per cent. |
| Potash, | K₂O | 35.50 | “ |
| Soda, | Na₂O | 0.57 | “ |
| Lime, | CaO | 5.68 | “ |
| Magnesia, | MgO | 15.19 | “ |
| Sulfuric acid, | SO₃ | 3.90 | “ |
| Insoluble, | 0.69 | “ | |
| Carbon dioxid and undetermined, | 7.46 | “ | |
The cakes left after the expression of the oil from flaxseed and other oily seeds are also very rich in nitrogenous matters; but these residues are chiefly used for cattle-feeding and only the undigested portions of them pass into the manure. Cottonseed cake-meal is not so well suited for cattle-feeding as the others mentioned, because of the cholin and betaïn which it contains; often in sufficient quantities to render its use dangerous to young animals. The danger in feeding increases as the total quantity of the two bases and also as the relative quantity of cholin to betaïn, the former base being more poisonous than the latter. In a sample of the mixed bases prepared in this laboratory from cottonseed cake-meal the cholin amounted to 17.5 and the betaïn to 82.5 per cent of the whole.[125]
The nitrogen contained in these bases is also included in the total nitrogen found in the meal. The actual proteid value of the numbers obtained for nitrogen is therefore less than that obtained for the whole of the nitrogen by the quantity present as nitrogenous bases.
In the United States cottonseed cake-meal is used in large quantities as a direct fertilizer but not so extensively for mixing as some of the other sources of nitrogen. Its delicate yellow color serves to distinguish it at once from the other bodies used for similar purposes. No special mention need be made of other oil-cake residues. They are quite similar in their composition and uses, and manner of treatment and analysis to the cottonseed product.
147. Fish Scrap.—Certain species of fish, such as the menhaden, are valued more highly for their oil and refuse than for food purposes. But even where fish in large quantities are prepared for human food, there is a considerable quantity of waste matter which is valuable for [Pg 165] fertilizing purposes. The residue of fish from which the fat and oil have been extracted, is dried and ground for fertilizing uses. The fish scrap thus obtained is used extensively, especially on the Atlantic border of the United States, for furnishing the nitrogenous ingredient in mixed fertilizers, and also for direct application to the fields. In fish flesh deprived of oil and water, the content of phosphoric acid is about two and one-half per cent, while the proteid matter may amount to three-quarters of the whole.[126]
The use of fish for fertilizing purposes is not new. As early as 1621 the settlers at Plymouth were taught to fertilize their maize fields by Squanto, an Indian. According to Goode, the value of nitrogen derived from the menhaden alone was two million dollars in 1875.[127] In 1878 it is estimated that 200,000 tons of these fish were captured between Cape Henry and the Bay of Fundy. The use of fish scrap for nitrogenous fertilizing has, since then, become an established industry, and the analyst may well examine his samples for this source of nitrogen when they are manufactured at points on the Atlantic coast, in proximity to great fishing centers.
148. Dried Blood and Tankage.—The blood and débris from abattoirs afford abundant sources of nitrogen in a form easily oxidized by the microorganisms of the soil. Blood is prepared for use by simple drying and grinding. The intestines, scraps, and fragments of flesh resulting from trimming and cutting, are placed in tanks and steamed under pressure to remove the fat. The residue is dried and ground, forming the tankage of commerce. Dried blood is richer in proteid matter than any other substance in common use for fertilizing purposes. When in a perfectly dry state, it may contain as much as fourteen per cent of nitrogen, equivalent to nearly eighty-eight per cent of proteid or albuminoid matter. Tankage is less rich in nitrogen than dried blood, but still contains enough to make it a highly desirable constituent of manures. Naturally, it would vary more in its nitrogen content than dried blood.
149. Horn, Hoof, and Hair.—These bodies, although quite rich in nitrogen, are not well suited to fertilizing purposes on account of the extreme slowness of their decomposition. Their presence, therefore, [Pg 166] should be regarded in the nature of a fraud, because by the usual methods of analysis they show a high percentage of nitrogen, and therefore acquire a fictitious value. The relative value of the nitrogen in these bodies as compared with the more desirable forms, is given in paragraph 5.
150. Ammoniacal Nitrogen.—In ammonia compounds, nitrogen is used chiefly for fertilizing purposes as sulfate. The ideal nitrogenous fertilizer would be a combination of the ammoniacal and nitric nitrogen found in ammonium nitrate. The high cost of this substance excludes its use except for experimental purposes.
151. Nitrogen in Guanos.—The nitrogen in guanos may be found partly as organic, partly as ammoniacal, and partly as nitric nitrogen. The high manurial value of guanos and bat deposits in caves, is due not only to their phosphoric acid, but also to the fact that part of the nitrogen is immediately available, while a part becomes assimilable by nitrification during the growing season. The content of nitrogen in guanos is extremely variable, and depends largely on the climatic conditions to which the deposit has been subjected. The state in which it exists is also a variable one, but with a constant tendency to assume finally the nitric condition.
The well-known habits of birds in congregating in rookeries during the nights, and at certain seasons of the year, tend to bring into a common receptacle the nitrogenous matters which they have gathered and which are deposited in their excrement and in the decay of their bodies. The feathers of birds are particularly rich in nitrogen, and the nitrogenous content of the flesh of fowls is also high. The decay therefore, of remains of birds, especially if it take place largely excluded from the leaching of water, tends to accumulate vast deposits of nitrogenous matter. If the conditions in such deposits be favorable to the processes of nitrification, the whole of the nitrogen, or at least the larger part of it, which has been collected in this débris, becomes finally converted into nitric acid, and is found combined with appropriate bases as deposits of nitrates. The nitrates of the guano deposits, and of the deposits in caves, arise in this way. If these deposits be subject to moderate leaching, the nitrate may become [Pg 167] infiltered into the surrounding soil, making it very rich in this form of nitrogen. The beds and surrounding soils of caves are often found highly impregnated with nitrates.
While for our purpose, deposits of nitrates only are to be considered which are of sufficient value to bear transportation, yet much interest attaches to the formation of nitrates in the soil even when they are not of commercial importance.
In many soils of tropical regions not subject to heavy rainfalls, the accumulation of these nitrates is very great. Müntz and Marcano[128] have investigated many of these soils, to which attention was called first by Humboldt and Boussingault. They state that these soils are incomparably more rich in nitrates than the most fertile soils of Europe. The samples which they examined were collected from different parts of Venezuela and from the valleys of the Orinoco, as well as on the shore of the Sea of Antilles. The nitrated soils are very abundant in this region of South America, where they cover large surfaces. Their composition is variable, but in all of them calcium carbonate and phosphate are met with, and organic nitrogenous material. The nitric acid is found always combined with lime. In some of the soils as high as thirty per cent of calcium nitrate have been found. Nitrification of organic material takes place very rapidly the year round in this tropical region. These nitrated soils are everywhere abundant around caves, as described by Humboldt, which serve as the refuge of birds and bats. The nitrogenous matters, which come from the decay of the remains of these animals, form true deposits of guano, which are gradually spread around, and which, in contact with the limestone and with access of air, suffer complete nitrification with the fixation of the nitric acid by the lime.
Large quantities of this guano are also due to the débris of insects, fragments of elytra, scales of the wings of butterflies, etc., which are brought together in those places by the millions of cubic meters. The nitrification, which takes place in these deposits, has been found to extend its products to a distance of several kilometers through the soil. In some places the quantity of calcium nitrate is so great in the soils that they are converted into a plastic paste by this deliquescent salt. [Pg 168]
152. Nitric Nitrogen.—In its purer forms, and suited to manurial purposes, nitric acid exists in combination with sodium as a compound commonly known as Chile saltpeter.
The existence of these nitrate deposits has long been known.[129] The old Indian laws originally prohibited the collection of the salt, but nevertheless it was secretly collected and sold. Up to the year 1821, soda saltpeter was not known in Europe except as a laboratory product. About this time the naturalist, Mariano de Rivero, found on the Pacific coast, in the Province of Tarapacà, immense new deposits of the salt. Later the salt was found in equal abundance in the Territory of Antofagasta, and further to the south in the desert of Atacama, which forms the Department of Taltal.
At the present time the collection and export of saltpeter from Chile is a business of great importance. The largest export which has ever taken place in one year was in 1890, when the amount exported was 927,290,430 kilograms; of this quantity 642,506,985 kilograms were sent to England and 86,124,870 kilograms to the United States. Since that time the imports of this salt into the United States have largely increased.
According to Pissis[130] these deposits are of very ancient origin. This geologist is of the opinion that the nitrate deposits are the result of the decomposition of feldspathic rocks, the bases thus produced gradually becoming united with the nitric acid provided from the air.
According to the theory of Nöllner[131] the deposits are of more modern origin, and due to the decomposition of marine vegetation. Continuous solution of soils beneath the sea gives rise to the formation of great lakes of saturated water, in which occurs the development of much marine vegetation. On the evaporation of this water, due to geologic isolation, the decomposition of nitrogenous organic matter causes generation of nitric acid, which, coming in contact with the calcareous rocks, attacks them, forming calcium nitrate, which, in presence of sodium sulfate, gives rise to a double decomposition into sodium nitrate and calcium sulfate.
The fact that iodin is found in greater or less quantity in Chile saltpeter is one of the chief supports of this hypothesis of marine [Pg 169] origin, inasmuch as iodin is always found in sea plants, and not in terrestrial plants. Further than this, it must be taken into consideration that these deposits of sodium nitrate contain neither shells nor fossils, nor do they contain any calcium phosphate. The theory, therefore, that they are due to animal origin is scarcely tenable.
Lately extensive nitrate deposits have been discovered in the U. S. of Columbia.[132] These deposits have been found extending over thirty square miles and vary in thickness from one to ten feet. The visible supply is estimated at 7,372,800,000 tons, containing from 1.0 to 13.5 percent of nitrate. The deposits consist of a mixture of sodium nitrate, sodium chlorid, calcium sulfate, aluminum sulfate, and insoluble silica. It is thought that the amount of these deposits will almost equal those in Chile and Peru.
153. Classification of Methods.—In general there are three direct methods of determining the nitrogen content of fertilizers. First the nitrogen may be secured in a gaseous form and the volume thereof, under standard conditions, measured and the weight of nitrogen computed. This process is commonly known as the absolute method. Practically it has passed out of use in fertilizer work, or is practiced only as a check against new and untried methods, or on certain nitrogenous compounds which do not readily yield all their nitrogen by the other methods. The process, first perfected by Dumas, who has also given it his name, consists in the combustion of the nitrogenous body in an environment of copper oxid by which the nitrogen, by reason of its inertness, is left in a gaseous state after the oxidation of the other constituents; viz., carbon and hydrogen, originally present.
In the second class of methods the nitrogen is converted into ammonia which is absorbed by an excess of standard acid, the residue of which is determined by subsequent titration with a standard alkali. There are two distinct processes belonging to this class, in one of which ammonia is directly produced by dry combustion of an organic nitrogenous compound with an alkali, and in the other ammonium sulfate is produced [Pg 170] by moist combustion with sulfuric acid, and the salt thus formed is subsequently distilled with an alkali, and the free ammonia thus formed estimated as above described. Nitric nitrogen may also be reduced to ammonia by nascent hydrogen either in an acid or alkaline solution as described in volume first.
In the third class of determinations is included the estimation of nitric nitrogen by colorimetric methods as described in the first volume. These processes have little practical value in connection with the analyses of commercial fertilizers, but find their chief use in the detection and estimation of extremely minute quantities of nitrites and nitrates. In the following paragraphs will be given the standard methods for the determination of nitrogen in practical work with fertilizing materials and fertilizers.
154. Official Methods.—The methods adopted by the Association of Official Agricultural Chemists have been developed by more than ten years of co-operative work on the part of the leading agricultural chemists of the United States. These methods should be strictly followed in all essential points by all analysts in cases where comparison with other data are concerned. Future experience will doubtless improve the processes both in respect of accuracy and simplicity, but it must be granted that, as at present practiced, they give essentially accurate results.
155. Volumetric Estimation by Combustion with Copper Oxid.—This classical method of analysis is based on the supposition that by the combustion of a substance containing nitrogen in copper oxid and conducting the products of the oxidation over red-hot copper oxid and metallic copper, all of the nitrogen present in whatever form will be obtained in a free state and can subsequently be measured as a gas. The air originally present in all parts of the apparatus must first be removed either by a mercury pump or by carbon dioxid or by both together, the residual carbon dioxid being absorbed by a solution of caustic alkali. Great delicacy of manipulation is necessary to secure a perfect vacuum and as a rule a small quantity of gas may be measured other than nitrogen so that the results of the analyses are often a [Pg 171] trifle too high. The presence of another element associated with nitrogen, or the possible allotropic existence of that element, may also prove to be a disturbing factor in this long-practiced analytical process. For instance, if nitrogen be contaminated with another element, e. g., argon, of a greater density the commonly accepted weight of a liter of nitrogen is too great and tables of calculation based on that weight would give results too high.
First will be given the official method for this process, followed by a few simple variations thereof, as practiced in this laboratory.
156. The Official Volumetric Method.—This process may be used for nitrogen in any form of combination.[133]
The apparatus and reagents needed are as follows:
Combustion tube of best hard Bohemian glass, about sixty-six centimeters long and 12.7 millimeters internal diameter.
Azotometer of at least 100 cubic centimeters capacity, accurately calibrated.
Sprengel mercury air-pump.
Small paper scoop, easily made from stiff writing paper.
Coarse cupric oxid.—To be ignited and cooled before using.
Fine cupric oxid.—Prepared by pounding ordinary cupric oxid in a mortar.
Metallic copper.—Granulated copper, or fine copper gauze, reduced and cooled in a current of hydrogen.
Sodium bicarbonate.—Free from organic matter.
Caustic potash solution.—Make a supersaturated solution of caustic potash in hot water. When absorption of carbon dioxid, during the combustion, ceases to be prompt, the solution must be discarded.
Filling the tube.—Of ordinary commercial fertilizers take from one to two grams for analysis. In the case of highly nitrogenized substances the amount to be taken must be regulated by the amount of nitrogen estimated to be present. Fill the tube as follows: (1) About five centimeters of coarse cupric oxid: (2) Place on the small paper scoop enough of the fine cupric oxid to fill, after having been mixed with the substance to be analyzed, about ten centimeters of the tube; [Pg 172] pour on this the substance, rinsing the watch-glass with a little of the fine oxid, and mix thoroughly with a spatula; pour into the tube, rinsing the scoop with a little fine oxid: (3) About thirty centimeters of coarse cupric oxid: (4) About seven centimeters of metallic copper: (5) About six centimeters of coarse cupric oxid (anterior layer): (6) A small plug of asbestos: (7) From eight-tenths to one gram of sodium bicarbonate: (8) A large, loose plug of asbestos; place the tube in the furnace, leaving about two and five-tenths centimeters of it projecting; connect with the pump by a rubber stopper smeared with glycerol, taking care to make the connection perfectly tight.
Operation.—Exhaust the air from the tube by means of the pump. When a vacuum has been obtained allow the flow of mercury to continue; light the gas under that part of the tube containing the metallic copper, the anterior layer of cupric oxid (see (5) above), and the sodium bicarbonate. As soon as the vacuum is destroyed and the apparatus filled with carbon dioxid, shut off the flow of mercury and at once introduce the delivery-tube of the pump into the receiving arm of the azotometer just below the surface of the mercury seal, so that the escaping bubbles will pass into the air and not into the tube, thus avoiding the useless saturation of the caustic potash solution.
Set the pump in motion and when the flow of carbon dioxid has very nearly or completely ceased, pass the delivery-tube down into the receiving arm, so that the bubbles will escape into the azotometer. Light the gas under the thirty centimeter layer of oxid, heat gently for a few moments to drive out any moisture that may be present, and bring to a red heat. Heat gradually the mixture of substance and oxid, lighting one jet at a time. Avoid a too rapid evolution of bubbles, which should be allowed to escape at the rate of about one per second or a little faster.
When the jets under the mixture have all been turned on, light the gas under the layer of oxid at the end of the tube. When the evolution of gas has ceased, turn out all the lights except those under the metallic copper and anterior layer of oxid, and allow to cool for a few moments. Exhaust with the pump and remove the azotometer before the flow of mercury is stopped. Break the connection of the tube with the pump, stop [Pg 173] the flow of mercury, and extinguish the lights. Allow the azotometer to stand for at least an hour, or cool with a stream of water until a permanent volume and temperature have been reached.
Adjust accurately the level of the potash solution in the bulb to that in the azotometer; note the volume of gas, temperature, and height of barometer; make calculation as usual, or read results from tables.
156. Note on Official Volumetric Method.—The determination of nitrogen in its gaseous state by combustion with copper oxid, has practically gone out of use as an analytical method. The official chemists rarely use it even for control work on samples sent out for comparative analysis. The method recommended differs considerably from the process of Jenkins and Johnson, on which it is based. The only source of oxygen in the official method is in the copper oxid. Hence it is necessary that the oxid in immediate contact with the organic matter be in a sufficiently fine state of subdivision, and that the substance itself be very finely powdered and intimately mixed with the oxidizing material. Failure to attend to these precautions will be followed by an incomplete combustion and a consequent deficit in the volume of nitrogen obtained.
The copper oxid before using is ignited, and is best filled into the tube while still warm by means of a long pointed metal scoop, or other convenient method. The copper spiral, after use, is reduced at a red heat in a current of hydrogen, and may thus be used many times.
157. The Pump.—Any form of mercury pump which will secure a complete vacuum may be used. A most excellent one can be arranged in any laboratory at a very small expense. The pump used in this laboratory for many years answers every purpose, and costs practically nothing, being made out of old material not very valuable for other use.
The construction of the pump and its use in connection with the combustion tube will be clearly understood from the following description:

Figure. 10.
Mercury Pump and
Azotometer.
A glass bulb I is attached, by means of a heavy rubber tube carrying a screw clamp, to the glass tube A, having heavy walls and a small [Pg 174] internal diameter, and being one meter or more in length. The tube A is continued in the form of a U, the two arms being joined by very heavy rubber tubing securely wired. The ends of the glass tubes in the rubber should be bent so that they come near together and form the bend of the U, the rubber simply holding them in place. This is better then to have the tube continuous, avoiding danger of breaking. A tee tube, T, made of the same kind of glass as A, is connected by one arm, a, with the manometer B, by a heavy rubber union well wired. The union is made perfectly air-tight by the tube filled with mercury held by a rubber stopper. The middle arm of the tee, a′, is expanded into a bulb, E, branching into two arms, one of which is connected with A and the other with the delivery-tube F, by the mercury-rubber unions, MM′, just described. The interior of the bulb E should be of such a shape as to allow each drop of mercury to fall at once into F without accumulating in large quantity and being discharged in mass. The third arm of the tee a″ is bent upwards at the end and passes into a mercury sealing tube, D, where it is connected by means of a rubber tube with the delivery-tube from the furnace. The flow of the mercury is regulated by the clamp C, and care should be taken that the supply does not get so low in I as to permit air bubbles to enter A. The manometer B [Pg 175] dips into the tube of mercury H. A pump thus constructed is simple, flexible, and perfectly tight. The only part which needs to be specially made is the tee and the one in use here was blown in our own laboratory. The bent end of the delivery-tube F may also be united to the main tube by a rubber joint thus aiding in inserting it into the V-shaped nozzle of the azotometer.
The azotometer used is the one devised by Schiff and modified by Johnson and Jenkins.[134]
We prefer to get the V nozzles separately and join them to any good burette by a rubber tube. The water-jacket is not necessary, but the apparatus can be left exposed until it reaches room temperature.
Any form of mercury pump capable of securing a vacuum may be used, but the one just described is commended by simplicity, economy, effectiveness, and long use.
158. The Pump and Combustion Furnace.—The pump and combustion furnace, as used in the laboratory, are shown in Fig. 10. The pump is constructed as just described, and rests in a wooden tray which catches and holds any mercury which may be spilled. The furnace is placed under a hood which carries off the products of the burning lamps and the hot air. A well-ventilated hood is an important accessory to this process, especially when it is carried on in summer. A small mercury pneumatic trough catches the overflow from the pump and also serves to immerse the end of the delivery-tube during the exhaustion of the combustion tube.
The other details of the arrangement and connections have been sufficiently shown in the previous paragraph.
159. Volumetric Method in this Laboratory.—It has been found convenient here to vary slightly the method of the official chemists in the following respects: The tube used for the combustion is made of hard refractory glass, which will keep its shape at a high red heat. It is drawn out and sealed at one end after being well cleaned and dried. It should be about eighty centimeters in length and from twelve to fourteen millimeters in internal diameter. The relative lengths of the spaces occupied by the several contents of the tube are approximately [Pg 176] as follows: Sodium bicarbonate, two; asbestos, three; coarse copper oxid, eight; fine copper oxid, containing sample, sixteen; coarse copper oxid, twenty-five; spiral copper gauze, ten to fifteen; copper oxid, eight; and asbestos plug, five centimeters, respectively.
The copper oxid should be heated for a considerable time to redness in a muffle with free access of air before using and the copper gauze be reduced to pure metallic copper in a current of hydrogen at a low red heat. The anterior layer of copper oxid serves to oxidize any hydrogen that may have been occluded by the copper. When a sample is burned containing all or a considerable part of the nitrogen as nitrates, the longer piece of copper gauze is used.
160. The Combustion.—The tube having been charged and connected with the pump it is first freed from air by running the pump until the mercury no longer rises in the manometer. The end of the tube containing the sodium bicarbonate is then gently heated so that the evolution of carbon dioxid will be at such a rate as to slowly depress the mercury in the manometer, but never fast enough to exceed the capacity of the pump to remove it. The lamp is extinguished under the sodium carbonate and the carbon dioxid completely removed by means of the pump. The delivery-tube is then connected with the azotometer, and the combustion tube carefully heated from the front end backwards, the copper gauze and coarse copper oxid being raised to a red heat before the part containing the sample is reached. When the nitrogen begins to come off, its flow should be so regulated by means of the lamps under the tube, as to be regular and not too rapid. From half an hour to an hour should be employed in completing the combustion. Since most samples of fertilizer contain organic matter, the nitrogen will be mixed with aqueous vapor and carbon dioxid. The former is condensed before reaching the azotometer, and the latter is absorbed by the potassium hydroxid. When the sample is wholly of a mineral nature it should be mixed with some pure sugar, about half a gram, before being placed in the tube. When bubbles of gas no longer come over, the heat should be carried back until there is a gradual evolution of carbon dioxid under the conditions above noted. Finally the gas is turned off [Pg 177] and the pump kept in operation until the manometer again shows a perfect vacuum when the operation may be considered finished. In the manipulation our chief variation from the official method consists in connecting the combustion apparatus with the measuring tube before the heat is applied to the front end of the combustion tube. Any particles of the sample which may have stuck to the sides of the tube on filling will thus be subject to combustion and the gases produced measured. Where it is certain that no such adhesion has taken place it is somewhat safer on account of the possible presence of occluded gases to heat the front end of the tube before connecting the combustion apparatus with the azotometer.
161. Method of Johnson and Jenkins.—In the method of Johnson and Jenkins the principal variation from the process described consists in introducing into the combustion tube a source of oxygen whereby any difficultly combustible carbon may be easily oxidized and all the nitrogen be more certainly set free.[135] The potassium chlorate used for this purpose is placed in the posterior part of the tube, which is bent at slight angle to receive it. The sodium bicarbonate is placed in the anterior end of the tube. The combustion goes on as already described, and at its close the potassium chlorate is heated to evolve the oxygen. The free oxygen is absorbed by the reduced copper oxid, or consumed by the unburned carbon. Any excess of oxygen is recognized at once by its action on the copper spiral. As soon as this shows signs of oxidation the evolution of the gas is stopped. Care must be taken not to allow the oxygen to come off so rapidly as to escape entire absorption by the contents of the combustion tube. In such a case the nitrogen in the measuring tube would be contaminated.
It is rarely necessary in fertilizer analysis to have need of more oxygen than is contained in the copper oxid powder in contact with the sample during the progress of combustion.
162. Calculation of Results.—The nitrogen originally present in a definite weight of any substance having been obtained in a gaseous form its volume is read directly in the burette in which it is collected. This instrument may be of many forms but the essential feature of its construction is that it should be accurately calibrated; and the divisions so graduated as to permit of the reading of the [Pg 178] volume accurately to a tenth of a cubic centimeter. For this purpose it is best that the internal diameter of the measuring tube be rather small so that at least each ten cubic centimeters occupies a space ten centimeters long. The volume occupied by any gas varies directly with the temperature and inversely with the pressure to which it is subjected. The quantity of aqueous vapor which a moist gas may contain is also a factor to be considered. Inasmuch as the nitrogen in the above process of analysis is collected over a strong solution of potassium hydroxid capable of practically keeping the gas in a dry state the tension of the aqueous vapor may be neglected.
163. Reading the Barometer.—Nearly all the barometers in use in this country have the scale divided in inches and the thermometers thereunto attached are graduated in Fahrenheit degrees. This is especially true of the barometers of the Weather Bureau which are the most reliable and most easy of access to analysts. It is not necessary to correct the reading of the barometer for altitude, but it is important to take account of the temperature at the time of observation. There is not space here to give minute directions for using a barometer. Such directions have been prepared by the Weather Bureau and those desiring it can get copies of the circular.[136]
The temperature of a barometer affects its accuracy in two ways. First the metal scale expands and contracts with changing temperatures: Second, the mercury expands and contracts also at a much greater rate than the scale. If a barometer tube hold thirty cubic inches of mercury the contents will be one ounce lighter at 80° F. than at 32° F. The true pressure of the air is therefore not shown by the observed height of the mercurial column unless the temperature of the scale and of the mercurial column be considered.
Tables of correction for temperature are computed by simple formulas based on the known coefficients of expansion of mercury and brass. For barometers with brass scales the following formula is used for making the correction:
| C = -h | t - 28.63 | . |
| 1.113t + 10978 |
In this formula t = temperature in degrees Fahrenheit and h = observed reading of the barometer in inches. [Pg 179]
Example:—Temperature observed 72°.5
Barometer reading observed, 29.415 inches,
from which C = 0.1165, and this number, according to the conditions of the formula, is to be subtracted from the observed reading. The true reading in the case given is, therefore,
| 29.298 inches or | 744.2 | millimeters. |
| The observed reading | 747.1 | “ |
| And the correction | 2.9 | “ |
Unless extremely accurate work be required the correction for temperature is of very little importance in nitrogen determinations in fertilizers. Each instrument sent out by the Weather Bureau is accompanied by a special card of corrections therefor, but these are of small importance in fertilizer work. In order then to get the correct weight of the gas from its volume the reading of the thermometer and barometer at the time of measurement must be carefully noted. However, after the end of the combustion, the azotometer should be carried into another room which has not been affected by the combustion and allowed to stand until it has reached the room temperature.
Every true gas changes its volume under varying temperatures at the same rate and this rate is the coefficient of gaseous expansion. For one degree of temperature it amounts to 0.003665 of its volume. Representing the coefficient of expansion by K the volume of the gas as read by V, the volume desired at any temperature by V′, the temperature at which the volume is read by t and the desired temperature by t′, the change in volume may be calculated by the following formula:
V′ = V[1 + K(t′ - t)].
Example.—Let the volume of nitrogen obtained by combustion be thirty-five cubic centimeters, and the temperature of observation 22°. What would be the volume of the gas at 0°?
Making the proper substitutions in the formula the equation is reduced to the form below:
V′ = 35[1 + 0.003665(0°-22°)]
or V′ = 35(1 - 0.08063) = 32.18.
Thirty-five cubic centimeters of nitrogen therefore measured at 22° become 32.18 cubic centimeters when measured at 0°. [Pg 180]
When gases are to be converted into weight after having been determined by volume, their volume at 0° must first be determined; but this volume must also be calculated to some definite barometric pressure. By common consent this pressure has been taken as that exerted by a column of mercury 760 millimeters in height. Since the volume of a gas is inversely proportional to the pressure to which it is subjected, the calculation is made according to that simple formula. Let the reading of the barometer, at the time of taking the volume of gas, be H, and any other pressure desired H′. Then we have the general formula:
| V : V′ = H′ : H; and V′ = | HV | . |
| H′ |
Example: Let the corrected reading of the barometer at the time of noting the volume of the gas be 740 millimeters, and the volume of the gas reduced to 0° be 32.18 cubic centimeters. What will this volume be at a pressure of 760 millimeters?
Substituting the proper values in the formula, we have:
| V′ = | (32.18 × 740) | = 31.33 |
| 760 |
Therefore, a volume of nitrogen which occupies a space of thirty-five cubic centimeters at a temperature of 22°, and at a barometric pressure of 740 millimeters, becomes 31.33 cubic centimeters at a temperature of 0° and a pressure of 760 millimeters.
One liter of nitrogen at 0° and 760 millimeters pressure weighs 1.25456 grams; and one cubic centimeter therefore 0.00125456 gram. To find the weight of gas obtained in the above supposed analysis, it will only be necessary to multiply this number by the volume of nitrogen expressed in cubic centimeters under the standard conditions; viz., 0.0125456 × 31.33 = 0.039305 gram. If the sample taken for analysis weighed half a gram, the percentage of nitrogen found would be 7.85.
164. Tension of the Aqueous Vapor.—It has been shown by experience that when a gas is collected over a potash solution containing fifty per cent of potassium hydroxid, the tension of the aqueous vapor is so far diminished as to be of no perceptible influence on the final result. To correct the volume of a gas, therefore, so collected for this tension, would involve an unnecessary calculation for practical purposes. If a gas thus collected should be transferred [Pg 181] to a burette over mercury, on which some water floats, then the correction should be made.
At 0° the tension of aqueous vapor will support a column of mercury 4.525 millimeters high, and at 40° one of 54.969 millimeters.
The following table gives the tension of aqueous vapors in millimeters of a mercurial column for each degree of temperature from zero to forty.
| Temperature. | Tension of vapor in millimeters. |
Temperature. | Tension of vapor in millimeters. |
|---|---|---|---|
| 0° | 4.525 | 21° | 18.505 |
| 1° | 4.867 | 22° | 19.675 |
| 2° | 5.231 | 23° | 20.909 |
| 3° | 5.619 | 24° | 22.211 |
| 4° | 6.032 | 25° | 23.582 |
| 5° | 6.471 | 26° | 25.026 |
| 6° | 6.939 | 27° | 26.547 |
| 7° | 7.436 | 28° | 28.148 |
| 8° | 7.964 | 29° | 29.832 |
| 9° | 8.525 | 30° | 31.602 |
| 10° | 9.126 | 31° | 33.464 |
| 11° | 9.751 | 32° | 35.419 |
| 12° | 10.421 | 33° | 37.473 |
| 13° | 11.130 | 34° | 39.630 |
| 14° | 11.882 | 35° | 41.893 |
| 15° | 12.677 | 36° | 44.268 |
| 16° | 13.519 | 37° | 46.758 |
| 17° | 14.409 | 38° | 49.368 |
| 18° | 15.351 | 39° | 52.103 |
| 19° | 16.345 | 40° | 54.969 |
| 20° | 17.396 |
When a gas is in contact with water the aqueous vapor is diffused throughout the mass, and the pressure to which the mixture is subjected, is partly neutralized by the tension of the water vapor. The real pressure to which the gas, whose volume is to be determined is subjected, is therefore diminished by that tension. If for instance a gas in contact with water show a volume of thirty-five cubic centimeters at 22° and 740 millimeters barometric pressure its volume is really greater than if it were perfectly dry. How much greater can be determined by inspecting the table, for at 22° the tension of water vapor is 19.675 millimeters of mercury. The real pressure to which the volume of gas is subjected is therefore 740 - 19.675 = 720.325 millimeters. [Pg 182]
If, therefore, in the example given, the nitrogen were in contact with water, the calculation would proceed as follows:
| V′ = | 32.18 × 720.325 | = 30.5 |
| 760 |
and 30.5 × 1.25456 = 38.26.
Hence, 38.26 milligrams of nitrogen correspond to 7.65 per cent, when half a gram of substance is taken for the combustion.
165. Aqueous Tension in Solutions of Potassium Hydroxid.—Even in strong solutions of potassium hydroxid the tension of aqueous vapor is not destroyed, but is reduced to a minimum, which is negligible in the calculation of the percentage by weight of the nitrogen in a sample of fertilizer. When dilute solutions of a caustic alkali are used however, the neglect of the tension of the aqueous vapor may cause an error of some magnitude. In such cases the strength of the solution should be known and correction made according to the following table:[137]
| Millimeters tension of aqueous vapor for KOH solutions of | |||||
|---|---|---|---|---|---|
| Temperature. | 9.09 per cent. |
16.66 per cent. |
23.08 per cent. |
28.57 per cent. |
32.89 per cent. |
| 10°.00 | 8.62 | 8.01 | 7.31 | 6.50 | 5.62 |
| 11°.00 | 9.21 | 8.56 | 7.82 | 6.95 | 6.01 |
| 12°.10 | 9.90 | 9.21 | 8.41 | 7.47 | 6.46 |
| 13°.00 | 10.50 | 9.77 | 8.92 | 7.93 | 6.86 |
| 13°.95 | 11.17 | 10.39 | 9.49 | 8.44 | 7.30 |
| 15°.15 | 12.06 | 11.22 | 10.25 | 9.11 | 7.86 |
| 16°.00 | 12.74 | 11.85 | 10.82 | 9.62 | 8.33 |
| 17°.00 | 13.57 | 12.63 | 11.54 | 10.26 | 8.88 |
| 18°.00 | 14.46 | 13.45 | 12.29 | 10.93 | 9.47 |
| 19°.00 | 15.39 | 14.33 | 13.09 | 11.65 | 10.09 |
| 20°.00 | 16.38 | 15.25 | 13.93 | 12.40 | 10.75 |
| 21°.00 | 17.42 | 16.22 | 14.82 | 13.20 | 11.44 |
| 21°.82 | 18.32 | 17.06 | 15.59 | 13.88 | 12.04 |
| 23°.00 | 19.68 | 18.32 | 16.75 | 14.92 | 12.94 |
| 24°.00 | 20.92 | 19.47 | 17.80 | 15.86 | 13.76 |
| 25°.00 | 22.19 | 20.67 | 18.91 | 16.85 | 14.62 |
| 26°.00 | 23.55 | 21.94 | 20.07 | 17.89 | 15.53 |
| 26°.98 | 24.95 | 23.25 | 21.27 | 18.96 | 16.46 |
| 27°.93 | 26.38 | 24.59 | 22.51 | 20.07 | 17.45 |
| 29°.00 | 28.08 | 26.18 | 23.96 | 21.38 | 18.59 |
| 30°.00 | 29.76 | 27.74 | 25.40 | 22.67 | 19.72 |
| 31°.00 | 31.51 | 29.38 | 26.91 | 24.03 | 20.91 |
| 32°.13 | 33.61 | 31.34 | 28.72 | 25.65 | 22.34 |
| 33°.00 | 35.30 | 32.93 | 30.18 | 26.97 | 23.50 |
| 34°.00 | 37.34 | 34.84 | 31.94 | 28.56 | 24.89 |
[Pg 183] 166. Use of Volumetric Method.—For practical purposes it may be said that the volumetric determination of nitrogen in fertilizer analysis has gone entirely out of use. For control and comparison it is still occasionally practiced but it has had to give way to the more speedy and fully as accurate processes of moist combustion with sulfuric acid which have come into general use in the last decade. The student and analyst however should not fail to master its details and become skilled in its use. There are certain nitrogenous substances such as the alkaloids which are quite refractory when subjected to moist combustion. While such bodies may not occur in fertilizers it is well to have at hand a means of accurately determining their nitrogen content.
167. Tables for Calculating Results.—Where many analyses are to be made by the copper oxid process it has proved convenient to shorten the work of calculating analyses by taking the data given in computation tables.[138] Before using these tables it must be known whether they are calculated on the supposition that the gas is measured in a moist state, partly moist, or wholly dry. Where the nitrogen is collected over water a table must be used in which allowance has been made for the tension of aqueous vapor. In case a saturated solution of a caustic alkali be used in the azotometer it is customary to take no account of the tension and the table employed must be constructed on this supposition. In point of fact even in the strongest alkali solution there is a certain amount of tension but this is so slight as only to affect the results in the second place of percentage decimals. Since, as a rule, only a few analyses are made by this method it will be found safer to use a caustic alkali solution of given strength and to calculate the results from the tables of aqueous tensions given above.
168. The Soda-Lime Process.—This process originally perfected by Varrentrap and Will, and improved by Peligot, was used almost exclusively by analysts until within the last decade for the determination of nitrogen not existing in the nitric form. It is based on the principle that when nitrogen exists as a salt of ammonia, or as an amid, or as proteid matter, it is converted into gaseous ammonia by combustion with an alkali. This ammonia can be carried into a set solution of acid by a stream of gas free of ammonia and the excess of [Pg 184] acid remaining after the combustion is complete can be determined by titration against a standard alkali solution. The results under proper conditions are accurate even when a small quantity of nitric nitrogen is present. When, however, there is any considerable quantity of this compound in the sample the method becomes inapplicable by reason of non-reduction of some of the nitrogen oxids produced by the combustion.
In bodies very rich in nitrogen such as urea all the nitrogen is not transformed directly into ammonia at the commencement of the combustion. A portion of it may unite with a part of the carbon to form cyanogen, which may unite with the soda to form sodium cyanid. With an excess of alkali, however, and prolonged combustion this product will be finally decomposed and all the nitrogen be secured as ammonia.
The nascent hydrogen which unites with the nascent nitrogen during the combustion is also derived from the organic matter which always contains enough carbon to decompose the water formed in order to be oxidized to carbon dioxid. While at first, therefore, during combustion, the hydrogen may unite with the oxygen, it becomes again free by the oxidation of the carbon and in this condition unites with the nascent nitrogen to form ammonia. In addition to carbon dioxid, ammonia, and free hydrogen there may also be found among the products of combustion marsh and olefiant gases and other hydrocarbon compounds which dilute, to a greater or less extent, the ammonia formed and help to carry it out of the combustion tube and into the standard acid.
169. The Official Method.—Reagents and Apparatus.—(1) Standard solutions and indicator the same as for the kjeldahl method:
(2) Dry granulated soda-lime, fine enough to pass a 2.5 millimeter sieve:
(3) Soda-lime, fine enough to pass a 1.25 millimeter sieve.[139]
Soda-lime may be easily and cheaply prepared by slaking two and one-half parts of quicklime with a strong solution of one part of commercial caustic soda, care being taken that there is enough water in the solution to slake the lime. The mixture is then dried and heated in an iron pot to incipient fusion, and, when cold, ground and sifted as above. [Pg 185]
Instead of soda-lime Johnson’s mixture of sodium and calcium carbonate, or slaked lime may be used. Slaked lime may be granulated by mixing it with a little water to form a thick mass, which is dried in the water-oven until hard and brittle. It is then ground and sifted as above. Slaked lime is much easier to work with than soda-lime, and gives excellent results, though it is probable that more of it should be used in proportion to the substance to be analyzed than is the case with soda-lime.
(4) Asbestos, which has been ignited and kept in a glass-stoppered bottle.
(5) Combustion tubes about forty centimeters long and twelve millimeters internal diameter, drawn out to a closed point at one end.
(6) Large-bulbed U tubes with glass stop-cock, or Will’s tubes with four bulbs.
Manipulation.—The substance to be analyzed should be powdered finely enough to pass through a sieve of one millimeter mesh; from seven-tenths to one and four-tenths grams, according to the amount of nitrogen present, are taken for the determination. Into the closed end of the combustion tube, put a small loose plug of asbestos, and upon it about four centimeters of fine soda-lime. In a porcelain dish or mortar, mix the substance to be analyzed, thoroughly but quickly, with enough fine soda-lime to fill about sixteen centimeters of the tube, or about forty times as much soda-lime as substance, and put the mixture into the combustion tube as quickly as possible by means of a wide-necked funnel, rinsing out the dish and funnel with a little more fine soda-lime, which is to be put in on top of the mixture. Fill the rest of the tube to within about five centimeters of the end with granulated soda-lime, making it as compact as possible by tapping the tube gently while held in a nearly upright position during the filling. The layer of granulated soda-lime should not be less than twelve centimeters long. Lastly, put in a plug of asbestos about two centimeters long, pressed rather tightly, and wipe out the end of the tube to free it from adhering soda-lime.
Connect the tube by means of a well-fitting rubber stopper or cork with the U tube or Will’s bulbs, containing ten cubic centimeters of standard acid, and adjust it in the combustion furnace so that the [Pg 186] end of the tube projects about four centimeters from the furnace, supporting the U tube or Will’s bulb suitably. Heat the portion of the tube containing the granulated soda-lime to a moderate redness, and when this is attained extend the heat gradually through the portion containing the substance, so as to keep up a moderate and regular flow of gases through the bulbs, maintaining the heat of the first part until the whole tube is heated uniformly to the same degree. Continue the combustion until gases have ceased bubbling through the acid in the bulbs, and the mixture of substance and soda-lime has become white, or nearly so, which shows that the combustion is finished. The process should occupy about three-quarters of an hour, or not more than one hour. Remove the heat, and when the tube has cooled below redness break off the closed tip and aspirate air slowly through the apparatus for two or three minutes to bring all the ammonia into the acid. Disconnect the tube, wash the acid into a beaker or flask, and titrate with the standard alkali.
During the combustion the end of the tube projecting from the furnace must be kept heated sufficiently to prevent the condensation of moisture, yet not enough to char the stopper. The heat may be regulated by a shield of tin slipped over the projecting end of the combustion tube.
It is found very advantageous to attach a bunsen valve to the exit tube, allowing the evolved gases to pass out freely, but preventing a violent sucking back in case of a sudden condensation of steam in the bulbs.
170. The Official French Method.—The French chemists prefer to drive out the traces of ammonia remaining in the combustion tube by means of the gases arising from the decomposition of oxalic acid.[140] The operation is conducted by mixing about one gram of oxalic acid with enough of dry granular soda-lime to form a layer of four centimeters in length at the bottom of the tube. The rest of the tube is then charged substantially as directed above. At the end of the combustion the oxalic acid is decomposed by heat furnishing sufficient hydrogen to remove from the tube all traces of ammonia which it may contain. The French chemists employ, for titration, either normal acids and alkalies or some decimal thereof or else an acid of such strength as to have each cubic centimeter thereof correspond to ten milligrams of nitrogen, [Pg 187] thus making the computation of results exceedingly simple. Such an acid is secured when one liter thereof contains thirty-five grams of pure monohydric sulfuric acid or forty-five grams of pure crystallized oxalic acid. The corresponding alkaline reagent should contain, in each liter, forty grams of pure potassium hydroxid.
171. The Hydrogen Method.—Thibault and Wagner recommend that the combustion with soda-lime be conducted in an atmosphere of hydrogen,[141] and Loges replaces this by common illuminating gas freed from ammonia by conducting it through a tube filled with glass balls moistened with dilute sulfuric acid.[142]
In these cases the combustion tube is left open at both ends and the materials under the tube confined to the proper position by asbestos plugs. The gases used act in a merely mechanical manner and their use affords so few advantages over the method of aspirating air at the end of the combustion as to render it unadvisable.
172. Coloration of the Product.—It often happens, especially in the combustion of animal products, such as tankage and fish scrap, that the acid securing the ammonia is deeply colored by the condensation of some of the other products of combustion. This coloration interferes in a very serious way with the delicacy of the indicator used to determine the end of the reaction. In this case the liquid may be mixed with an alkali and distilled and the ammonia secured in a fresh portion of the standard acid as in the moist combustion process to be hereafter described.
173. General Considerations.—(1) Preparation of Sample.—In the soda-lime method it is of great importance that the organic substances be in a fine state of subdivision so as to admit of intimate mixture with the alkali. In cases where fragments of hoof, horn, hair, or similar substances are to be prepared for combustion it is advisable to first decompose them by heating with a small quantity of sulfuric acid. The excess of acid may be neutralized with marble dust and the resulting mixture dried, rubbed to a fine powder, and mixed with the soda-lime in the usual way. Care must be taken not to lose any of the ammonia from the sulfate which may be formed in mixing with the soda-lime in filling the tube. [Pg 188]
(2) Purity of Soda-Lime.—The soda-lime employed must be entirely free of nitrogenous compounds and some blank combustions should be made in proof of its purity.
(3) Temperature.—The temperature of the combustion should not be allowed to exceed low redness. At very high temperatures there would be danger of decomposing the ammonia.
(4) Aspiration of Air.—Before aspiring a current of air through the tube to remove the last traces of ammonia the gas should be put out under the furnace and the tube be allowed to cool below redness to avoid any danger of acting on the nitrogen in the air.
174. The Ruffle Soda-Lime Method.—Many attempts have been made to adapt the soda-lime method to the determination of nitric nitrogen. Of these the process devised by Ruffle is the only one which has proved successful.[143] The method is founded on the action of sulfurous vapors on the nitrogen oxids produced during the combustion whereby sulfuric acid is formed and the nascent nitrogen is joined with hydrogen to form ammonia. By this process all the nitrogen contained in the sample, even if in the nitric form, is finally obtained as ammonia. In the original method the reagents employed were a mixture of sodium thiosulfate and soda-lime and a mixture of charcoal, sulfur, and granulated soda-lime. Subsequently the official chemists substituted sugar for the charcoal.[144] The method was used for a long time by the official chemists and came into general favor until displaced by the simpler and cheaper processes of the moist combustion method adapted to nitric nitrogen. As finally modified and used by the official chemists the process was conducted as described below.
175. The Official Ruffle Method.—[145]Reagents.—(1) Standard solutions and indicator the same as for the kjeldahl method.
(2) A mixture of equal parts by weight of fine-slaked lime and finely powdered sodium thiosulfate dried at 100°:
(3) A mixture of equal parts of weight of finely powdered granulated sugar and flowers of sulfur:
(4) Granulated soda-lime, as described under the soda-lime method: [Pg 189]
(5) Combustion tubes of hard Bohemian glass seventy centimeters long and one and three-tenths centimeters in diameter:
(6) Bulbed U tubes or Will’s bulbs, as described under the soda-lime method:
Manipulation.—(a) Clean the U tube and introduce ten cubic centimeters of standard acid.
(b) Fit the cork and glass connecting tube. Fill the tube as follows: (1) A loosely fitting plug of asbestos, previously ignited, and then two and five-tenths to three and five-tenths centimeters of the thiosulfate mixture: (2) The weighed portion of the substance to be analyzed is intimately mixed with from five to ten grams of the sugar and sulfur mixture: (3) Pour on a piece of glazed paper or in a porcelain mortar a sufficient quantity of thiosulfate mixture to fill about twenty-five centimeters of the tube; then add the substance to be analyzed, as previously prepared, mix carefully, and pour into the tube; shake down the contents of the tube; rinse off the paper or mortar with a small quantity of the thiosulfate mixture and pour into the tube; then fill up with soda-lime to within five centimeters of the end of the tube: (4) Place another plug of ignited asbestos at the end of the tube and close with a cork: (5) Hold the tube in a horizontal position and tap on the table until there is a gas-channel along the top of the tube: (6) Make connection with the U tube containing the acid; aspirate and see that the apparatus is tight.
The Combustion.—Place the prepared combustion tube in the furnace, letting the open end project a little, so as not to burn the cork. Commence by heating the soda-lime portion until it is brought to a full red heat. Then turn on slowly jet after jet toward the outer end of the tube, so that the bubbles come off two or three a second. When the whole tube is red hot and the evolution of the gas has ceased and the liquid in the U tube begins to recede toward the furnace, attach the aspirator to the other limb of the U tube, break off the end of the tube, and draw a current of air through for a few minutes. Detach the U tube and wash the contents into a beaker or porcelain dish; add a few drops of the cochineal solution, and titrate.
176. Observations.—In our experience we have found it much more satisfactory to adhere to the earlier directions for preparing the [Pg 190] mixture of thiosulfate and alkali. We much prefer to make the mixture with soda-lime and without the previous drying of the sodium salt. Ruffle himself says that the sodium thiosulfate should be dry but not deprived of its water of crystallization.[146] The best method to dry the crystal powder without depriving it of its crystal water is to press it between blotting papers. The official method also contains a typographical error in prescribing that the combustion tube should have a length of thirty centimeters where evidently thirty inches were meant. Ruffle’s original tube was twenty-two inches in length.
As is seen from the above description the method is essentially a reduction process by the action of a powerful deoxidizer in the presence of an alkali. The crystals of the thiosulfate salt cannot be brought into direct contact with a pure alkali, like soda or potash, without forming at once a wet mass which would tend to cake and obstruct the tube. The soda-lime is therefore a mechanical device to prevent this fusion. Where many analyses are to be made an iron tube, for economical reasons, may be substituted for the glass; but the glass tube permits a more intelligent observation of the progress of the analysis.
Since charcoal has very high absorbent powers it will be found always to contain a little nitrogen which may be in a form to generate ammonia during the combustion. The charcoal used should therefore be previously boiled with caustic soda or potash solution, dried, powdered, and preserved in well-stoppered bottles. Although pure sugar is practically free of nitrogen, even when it is used, it is advisable to occasionally make a blank determination and thus ascertain the correction to be made for possible contamination.
177. Boyer’s Modification of Ruffle’s Method.—The principle of the method rests on the observation that if nitrates be heated in a combustion tube with calcium oxalate and soda-lime, not more than two-thirds of the total nitrogen appear as ammonia; but if a certain proportion of sulfur be added the whole of the nitrogen is recovered.[23] The process may be divided into two reactions; viz.:
(1) Action of the calcium oxalate upon the sodium nitrate in presence of soda-lime: [Pg 191]
(2) The action of sulfurous acid and of calcium oxalate upon the sodium nitrate in presence of soda-lime.
The analysis is conducted as follows: Dry and pulverize one-half gram of nitrate (Na or K) and mix it intimately with fifty grams of the reducing compound containing approximately ten per cent sulfur, 22.5 per cent neutral calcium oxalate, and 67.5 per cent soda-lime. The combustion tube is charged as follows:
Length of tube fifty-five centimeters:
Diameter of tube seventeen millimeters:
Add first two grams pulverized calcium oxalate:
Add next ten grams pulverized soda-lime:
“ “ ten grams of the reducing compound:
“ “ the nitrate incorporated with fifty grams of the reducing mixture:
Add next ten grams of the reducing mixture:
“ “ ten grams pulverized soda-lime:
The tube is then lightly closed with an asbestos plug.
The tube is heated gradually from the front backwards, the calcium oxalate furnishing finally the gas necessary to drive out the last traces of ammonia. The process is equally applicable to the determination of nitrogen in all its forms or to mixtures thereof.
The method has also been applied to the mixture of ammoniacal and organic nitrogen and to the mixture of ammoniacal, nitric, and organic nitrogen, the combustions having been made both in an iron and a glass tube. The amounts of material to be used vary from one-half gram to a gram, according to its richness in nitrogen.
The combustion should be terminated in forty minutes.
When a combustion is terminated, the acid containing the ammonia is placed in a beaker and boiled for two or three minutes to drive off the sulfurous and carbonic acids. The titration is then conducted in the usual manner.
The combustion can be carried on just as well in an iron tube as in a glass one. The reagents employed, especially soda-lime, being hygroscopic, a little water is disengaged in heating, which is condensed at the cold extremity of the tube, and which may absorb a little ammonia, unless special precautions are taken to have the materials dry. [Pg 192]
178. Historical.—As long ago as 1868 Wanklyn proposed to conduct the combustion of organic bodies in a wet way, using potassium permanganate as the oxidizing body.[147] About ten years after this he attempted to extend the method so as to estimate the quantity of proteid matter in a sample by treatment with an alkaline solution in presence of the permanganate salt. One gram of the finely pulverized sample was treated in a liter flask with one-tenth normal potash lye. After digestion for some time, from ten to twenty cubic centimeters were taken for the determination. According to the supposition of Wanklyn, pure albuminoid matters thus treated yielded one-tenth of their weight of ammonia, or about fifty per cent of the total nitrogen appeared as ammonia. The ammonia content of the sample was determined by the colorimetric process devised by Nessler. It is needless to add that the process of Wanklyn proved to be of no practical use whatever, acting differently on different albuminoid matters, and even on the same substance. No other attempt was made to perfect the moist combustion process until Kjeldahl[148] introduced the sulfuric acid method in 1883. The simplicity, economy, and adaptability of this method have brought it into general use. At first the process was only applied to organic nitrogenous compounds in the absence of nitrates, but especially by the modifications proposed by Asboth, Jodlbaur, and Scovell, it has been made applicable to all cases, with the possible exception of a few alkaloidal and allied bodies. The moist combustion process for determining nitrogen is now generally employed by chemists in all countries, not only for fertilizer control, but also for general work.
179. The Method of Kjeldahl.—The process originally proposed by Kjeldahl is applicable only to nitrogenous bodies free of nitric nitrogen. The principle of the process is based on the action of concentrated sulfuric acid at the boiling-point in decomposing nitrogenous compounds without producing volatile combinations and the subsequent completion of the oxidation by means of potassium permanganate. The original process has been modified by many analysts [Pg 193] but the basic principle of it has remained unchanged. It will therefore prove useful here to describe the process as originally given.[149]
The weighed substance is placed in a small flask. With solid bodies this is a very simple operation, but with liquids more difficult. Liquids which are not decomposed, on heating, should be evaporated in a thin glass dish which can be ground up and placed in the digestion flask with the desiccated sample. The strongest sulfuric acid is added in sufficient quantity to secure complete decomposition, not less than ten cubic centimeters in any case. The acid must be free of ammonia and be kept in such a way as not to absorb ammonia from the atmosphere of the laboratory. To guard against danger of error from such an impurity frequent control determinations should be made. In control experiments one or two grams of pure sugar should be used as the organic matter. If the acid employed contain traces of ammonia the necessary corrections should be made in each analysis. The flask having been charged is placed on a wire gauze over a small flame. The organic matter becomes black and tar-like and soon there is a rapid decomposition attended with the evolution of gaseous products among which sulfur dioxid is found. To avoid danger from spurting, the digestion flask should be placed in an oblique position. The flask should have, at least, a capacity of 100 cubic centimeters and a long neck and be made of a kind of glass capable of withstanding the action of the boiling acid. Particles of the carbonized organic matter left on the sides of the flask by the foaming of the mass at first are gradually dissolved by the vapors of the boiling acid as the digestion proceeds. The action of the sulfuric acid is not entirely finished when gases cease to be given off but the digestion should be continued until the liquid in the flask is clear and colorless or nearly so. Usually about two hours are required to secure this result. When aided by the means mentioned below the time of digestion can be very materially shortened. By adding some fuming sulfuric acid, or glacial phosphoric acid, the dilution caused by the formation of water in the combustion of the organic matter, can be avoided. For albuminoid bodies it is hardly necessary to continue the combustion until all carbonaceous matter is destroyed. The full [Pg 194] complement of ammonia is usually obtained after an hour’s combustion even if the liquid be still black or brown, but with other nitrogenous bodies the case is different so that upon the whole it is safest to secure complete decoloration.
The temperature must be maintained at the boiling-point of the acid or near thereto since at a lower temperature, for instance from 100° to 150°, the formation of ammonia is incomplete. Since all organic substances of whatever kind are dissolved by the boiling acid the previous pulverization of the material need be carried only far enough to secure a fair sample. Many substances give up practically all their nitrogen as ammonium sulfate when heated with sulfuric acid as, for instance, urea, asparagin, and the glutens. In most of the other organic bodies fully ninety per cent of the nitrogen are likewise secured as the ammonium salt. In the aromatic compounds, or even in the form of amid in anilin salts, the nitrogen is more resistant to the action of sulfuric acid. In the alkaloids where the nitrogen is probably a real component of the benzol skeleton the formation of ammonia is very incomplete. But even in the cases where the conversion of the nitrogen into ammonia is practically perfect it is advisable to finish the process by completing the oxidation with potassium permanganate. The permanganate should be used in a dry powdered form and added little by little to the hot contents of the digestion flask, the latter being held in an upright position and removed meanwhile from the lamp. When carefully performed there is no danger of loss of ammonia although the oxidation is, at times, so vigorous as to be attended with evolution of light. The permanganate must always be added in excess and until a permanent green color is produced. The flask is then gently heated for from five to ten minutes over a small flame, but this is not important. The heating must not be too strong or else a strong evolution of oxygen will take place with a consequent reduction of the manganese compound. When this happens the liquid again becomes clear and there is a loss of ammonia.
After cooling, the contents of the flask are diluted with water, the green color giving place to a brown with a rise of temperature. After cooling a second time the whole is brought into a distillation flask of about three-quarters of a liter capacity and attached to a condenser [Pg 195] which ends in a vessel containing titrated sulfuric acid. About forty cubic centimeters of sodium hydroxid solution of one and three-tenths specific gravity are added and the stopper at once inserted to prevent any loss of ammonia. To prevent bumping during the distillation some zinc dust is added securing an evolution of hydrogen during the progress of the distillation. In this case the bumping is prevented until near the end of the operation when it begins anew, probably by reason of the separation of solid sodium sulfate. After the end of the distillation, the excess of acid remaining in the receiver is determined by a set alkali solution and thus the quantity of ammonia obtained easily calculated. Kjeldahl, however, preferred to titrate the solution after adding potassium iodate and iodid, a mixture which in the presence of a strong acid sets free a quantity of iodin equivalent to the free acid present. The iodin thus set free is titrated by a set solution of sodium thiosulfate using starch as an indicator. The merits of this method are sharpness of the end reaction and the possibility of using only a small quantity of the nitrogenous body for the combustion. The sulfuric acid used in the receiver is made of the same strength as the thiosulfate solution; viz., about one-twentieth normal. Thirty cubic centimeters of this were found to be the proper amount for use with substances taken in such quantities as to produce ammonia enough to neutralize about half of it. The titration is carried on as follows: A few crystals of potassium iodid are dissolved in the acid mixture obtained after the distillation is completed, then a few drops of the starch-paste and finally a few drops of a four per cent solution of potassium iodate. The iodin set free is then oxidized by the addition of the one-twentieth normal sodium thiosulfate solution until the blue color disappears.
| Example: Sulfuric acid used, | 30 | cc. | ||
| Equivalent to sodium thiosulfate, | 30 | cc. | ||
| Blank combustion required, | 29.8 | cc. | thiosulfate | solution. |
| Combustion of 0.645 gram of barley required, | 14.5 | cc. | “ | “ |
| Thiosulfate corresponding to barley, | 15.3 | cc. |
[Pg 196] In the computation it is more simple to multiply the corresponding number of cubic centimeters of thiosulfate by seven, half the atomic weight of nitrogen, and divide the product by the weight of the substance taken, which will give the per cent of nitrogen therein.
| Then | 15.3 × 7 | × 100 = 1.66 = per cent of nitrogen in sample taken. |
| 0.645 |
A more detailed description of the method of making the titration follows: After the distillation is finished the condensing-tube is rinsed with a little water, after which the sulfuric acid unneutralized in the receiver is determined. It is advisable first to test the reaction of the distillate with litmus paper before going any further; for if at any time all the acid should be found neutralized it will be necessary to add a sufficient quantity of one-twentieth normal sulfuric acid before adding the potassium iodid, etc., otherwise the determination will be irreparably lost. Add to the contents of the flask ten cubic centimeters of the potassium iodid and two cubic centimeters of the potassium iodate solutions, described further on, and the sodium thiosulfate is then run in from a burette till the fluid, which is constantly kept agitated by shaking the flask, shows only a bare trace of yellow coloration from the iodin still present. Starch solution is then added, and the blue color obtained is at once removed by additional thiosulfate solution. When some experience has been gained, the eye is able to discern, with great certainty, even the slight coloration caused by only a small trace of free iodin.
In regard to the sensitiveness of the end reaction and the accuracy of the result, this method of titration leaves nothing to be wished for. The strength of the thiosulfate solution is determined in exactly the same manner, and with starch as an indicator. For this purpose, measure ten cubic centimeters of one-twentieth normal sulfuric acid into an erlenmeyer, add 120 cubic centimeters of ammonia-free water, ten cubic centimeters of potassium iodid solution, and two cubic centimeters of iodate solution; add thiosulfate solution till the fluid shows only the above-mentioned light yellow tint, then add starch, and finally thiosulfate. In this way the strength of the thiosulfate is [Pg 197] ascertained, which of course must be occasionally re-determined, under exactly the same conditions as with the nitrogen determinations themselves, and every possible error is thereby excluded. That the solution once decolorized within a short time again assumes a deep blue color, is a matter of no concern, inasmuch as both solutions are added in such a manner that the end reaction lies exactly at the point when the starch iodid reaction distinctly disappears.
180. Theory of the Reactions.—As has been seen above the final product of heating a nitrogenous organic compound with sulfuric acid and an oxidizing body is ammonium sulfate. The various steps by which this is obtained have been traced by Dafert:[150]
(1) The sulfuric acid abstracts from the organic matter the elements of water:
(2) The sulfur dioxid produced by the action of the residual carbon on sulfuric acid exercises a reducing effect on the nitrogenous bodies present:
(3) From the nitrogenous bodies produced by the above reduction ammonia is formed by the action of an oxidizing body:
(4) The ammonia formed is at once fixed by the acid as ammonium sulfate. According to the theory of Asboth the hydrogen which is formed during the action of sulfuric acid on organic matter, when in a nascent state, also aids greatly in the production of ammonia. This idea is based on the fact that with those bodies which afford a deficit of hydrogen the formation of ammonia is imperfect.[151]
181. Preparation of Reagents.—(1) Pure Sulfuric Acid.—As is well known the so-called pure sulfuric acid in the market usually contains ammonia, a fact which compelled Kjeldahl to determine the quantity of nitrogen in the acid in every instance, and to make correction for the same in the analysis. An acid absolutely free from this impurity may, however, easily be prepared by the distillation of the commercial article in a small glass retort holding easily about 400 cubic centimeters. To conduct this operation without danger it is only necessary to arrange the apparatus so that the heavy fluid is heated to boiling, not from the bottom of the retort, but from its sides, and that the upper portion of the body and neck is kept [Pg 198] sufficiently warm so that the sulfuric acid fumes are not allowed to condense and flow back into the retort. Both these ends are attained simply by surrounding the retort with a piece of sheet iron, cylinder-shaped beneath, and with an oval upper part, having an opening of about one centimeter in diameter for the neck of the retort. To conduct the distillation a burner is used with an arrangement for spreading the flame. To avoid with certainty all bumping of the sulfuric acid and the resulting danger therefrom, the lamp is so arranged that only the products of combustion go up between the retort and its iron hood, without allowing the flame itself to come into contact with the glass vessel. The retort should be filled about half full, or with 200 cubic centimeters of acid. By this device, without any danger whatever, about one liter of sulfuric acid may be distilled in a day. The retort will stand numerous distillations. Once begun, the distillation takes care of itself; it is necessary to discontinue it when only the bottom of the retort is covered with sulfuric acid, and to fill fresh acid through a funnel when the retort has cooled off. The first twenty cubic centimeters of the distillate going over are collected by themselves and rejected. What comes over later is, as shown by experience, absolutely ammonia-free, and can be used without any correction, for the nitrogen determinations according to Kjeldahl. The acid is kept in a stoppered bottle in a place not reached by ammonia fumes. The ten cubic centimeter pipette used for measuring the quantity of sulfuric acid required for each determination, is fastened in the perforated rubber stopper with which the bottle is kept closed, and is itself closed above by a small rubber tube with a plug of glass wool in it.
(2) Potassium Permanganate.—Crystals of this salt are crushed (not pulverized) with a pestle into small pieces of about one-half millimeter size, which are kept in a long glass tube of about ten millimeters diameter, closed with a stopper.
(3) Ammonia-free Water.—Common distilled water cannot be used in the determination of nitrogen according to Kjeldahl, since it contains ammonia. It may be obtained free from the same by redistillation in a large glass retort with the addition of a few drops of sulfuric acid. All vessels used in the determination are rinsed out beforehand with this water. [Pg 199]
(4) Ammonia-free Soda-lye is most conveniently prepared by adding 270 grams of common sodium hydroxid in sticks, little by little, to one liter of distilled water which is kept continually boiling, by means of a small flame, in a good-sized silver dish. The dish is kept covered with a glass plate. Care has to be exercised not to add the alkali too rapidly, nor in too large quantities at a time for in this case the fluid will boil too violently at every addition of the alkali. After the operation is finished the lye is at once siphoned into a glass flask, and when cold, is poured into a glass-stoppered bottle.
(5) One-twentieth Normal Sulfuric Acid is prepared from sulfuric acid and water both absolutely ammonia-free, and is kept in a place where no fumes of ammonia can reach it, in a well-stoppered glass bottle, the stopper being smeared with vaseline.
(6) Sodium Thiosulfate Solution.—This should be of the same strength as the one-twentieth normal sulfuric acid. It is prepared by dissolving the salt in ammonia-free water, and is compared with the acid, to which has been added potassium iodid and iodate, using starch as an indicator, in the manner described above. The solution is kept in a well-stoppered bottle, in the dark. When the salt and water used are perfectly pure, it will keep unchanged for a long time.
(7) Potassium Iodid.—Dissolve five grams of chemically pure potassium iodid in ammonia-free water and make the volume 100 cubic centimeters. Ten cubic centimeters of this solution are used for each determination; keep the solution in the dark and in a well-stoppered bottle.
(8) Potassium Iodate.—Dissolve four grams of chemically pure potassium iodate in ammonia-free water and make the volume 100 cubic centimeters. Use two cubic centimeters of this solution for each determination.
(9) Starch Solution.—Digest pure starch for about a week with dilute hydrochloric acid, wash perfectly free from chlorin by decantation, and finally dry it between filter-paper. The starch is then dissolved in water with the aid of heat. Such a solution will keep for an indefinite time, if it be saturated with common salt. Ten grams of this starch are dissolved in 1,000 cubic centimeters of ammonia-free water; use one or two cubic centimeters for each determination. [Pg 200]
182. Kjeldahl Method as Practiced by the Holland Royal Experiment Station.—Necessary Reagents: 1. Phosphosulfuric acid, made by mixing a liter of sulfuric acid of specific gravity 1.84 with 200 grams of phosphoric anhydrid:
2. Alkaline sodium sulfid solution, made by dissolving 500 grams of sodium hydroxid and six grams of sodium sulfid or eight and one-half grams of potassium sulfid, in a liter of water:
3. Mercury:
4. Paraffin in small pieces:
5. Dilute sulfuric acid and dilute potash solution, both of known strength:
6. Pieces of previously ignited pumice stone or of granulated zinc:
7. Neutral solution of rosolic acid or litmus.
Apparatus: The apparatus necessary consists of oxidation flasks of about 200 cubic centimeters capacity and distillation flasks of about 500 cubic centimeters capacity, both of bohemian glass. Copper may be used for the distillation flasks.[152]
The Process: A gram of the sample to be analyzed is placed in an oxidation flask together with twenty cubic centimeters of phosphosulfuric acid and a drop of mercury, about 600 milligrams, and heated till the fluid becomes colorless. After cooling, dilute and wash the contents of the flask into a distillation flask. The resulting volume should be about 300 cubic centimeters. Add 100 cubic centimeters of the alkaline sodium sulfid solution and some pieces of ignited pumice stone or granulated zinc. Distill the ammonia, receiving the distillate in a flask containing a known volume of the standard sulfuric acid. Titrate with tenth-normal potash, using litmus or rosolic acid as indicator.
183. The Kjeldahl Method as Practiced at the Halle Station.—The method at present in vogue in the German stations of conducting the moist combustion process is well illustrated by the method of procedure followed at Halle.[29] From seven-tenths to one and five-tenths grams of the sample are taken for analysis according to its richness in nitrogen. Because of the fact that so small a quantity of the sample is taken it is of the highest importance that it be perfectly homogeneous throughout its entire mass. Otherwise, grave errors may arise. From the [Pg 201] sample, as sent to the laboratory, the analyst should take a subsample and this should be rubbed to a fine powder and the part used for analysis carefully taken therefrom. If the sample be moist it may be rubbed up with an equal weight of gypsum, in which case a double quantity is taken for the determination. Substances like bone-meal, which do not keep well mixed, especially when occasionally shaken, should be intimately mixed before each weighing. The sample taken for analysis is placed in a glass flask of about 150 cubic centimeters capacity. The flasks should be made of a special glass to withstand the tension of the combustion. Those made by Kavalier at Sazava, in Bohemia, have proved to be the most lasting. A globule of mercury weighing a little less than one gram is placed in the flask and also twenty cubic centimeters of pure sulfuric acid of 1.845 specific gravity. The mercury is conveniently measured by an apparatus suggested by Wrampelmayer. It consists of an iron tube holding mercury, and is conveniently filled, from time to time, from a supply vessel placed in a higher position and joined by means of a heavy glass tube and rubber tube connections. The lower end of the iron tube is provided with a movable iron stopper having a pocket just large enough to hold a globule of mercury, weighing a little less than a gram. On turning the stopper the pocket is brought opposite a discharge orifice and the measured globule of mercury is discharged. With substances which tend to produce a strong foaming a little paraffin is used. The flasks after they are charged are placed on circular digesting ovens under a hood as shown in figure 11.
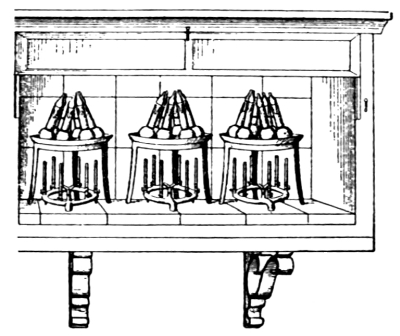
Figure. 11.
Moist Combustion Apparatus of the
Halle Agricultural Laboratory.
At first the tripodal support of the flasks is so turned as to bring them between the lamps and in this way a too rapid reaction is at first avoided. After half an hour the tripods are so turned as to bring each flask directly over the lamp, the flame of which is allowed to impinge directly against the glass. The flame is so regulated that after the [Pg 202] evolution of the sulfur dioxid has nearly ceased the contents of the flask are brought into gentle ebullition. The boiling is continued until the contents of the flask are colorless, usually about two hours. As a rule such substances as cottonseed-meal and dried blood will take a longer time for complete combustion than other fertilizing materials. During the combustion the flasks are closed with an oblong loose-fitting unground glass stopper. When the oxidation is finished the contents of the flasks are allowed to cool, the stoppers are removed, and enough water is added to fill the flasks about three-quarters full. The flasks are gently shaken, and the possibility of breaking, from the heat developed, must not be overlooked. To avoid confusion the flasks are all numbered before beginning the work, and the numbers noted by the analyst in connection with the samples. The contents of each one are next poured into the distillation flask and the digestion vessels are washed with 100 cubic centimeters of water in three portions, and the wash-water added to the liquid. Sometimes in washing out the digestion flask yellow basic mercury compounds separate on its walls, but this does not, in any way, influence the accuracy of the results. The distillation flasks should have about 600 cubic centimeters capacity. To avoid the transfer the digestion may take place in the distillation flask in which case the latter must be made of special glass as indicated.
To the liquid thus transferred are added seventy-five cubic centimeters of soda-lye containing one and one-half times as much potassium sulfid as is necessary to combine with the mercuric sulfate present. The lye is of such a strength that sixty cubic centimeters are sufficient to neutralize the acid present. It has a specific gravity of 1.375 and contains thirty-three grams of potassium sulfid in a liter.
In order to avoid the bumping which may take place during the distillation, some granulated zinc should be added.
The distillation flask is closed with a rubber stopper carrying a bulb-tube which ends above in a glass tube about three-quarters of a meter long, bent at an acute angle, and passing obliquely downward on a convenient support. This tube is connected by a rubber, with the end tube bent nearly at a right angle and dipping into the standardized [Pg 203] acid in the erlenmeyer receiver. The general arrangement of the distilling apparatus is shown in figure 12. Since the contents of the vessel are warmed by mixing with the soda-lye, the flame can be turned on at full head at once at the commencement of the operation. In about a quarter of an hour the liquid in the receiver will be at the boiling-point, and the boiling should be continued for five minutes more, making twenty minutes in all for the completion of the distillation. By this boiling the contents of the receiver are not charged with carbon dioxid, as might happen if a condenser were used. The receiver contains twenty cubic centimeters of a standardized sulfuric acid solution and about fifty cubic centimeters of water.
Figure. 12.
Distillation Apparatus of
Halle Agricultural Laboratory.
The acid used should contain 38.1 grams of sulfuric acid of 1.845 specific gravity in a liter; and it should be set by titration with chemically pure sodium carbonate. For this purpose seven-tenths gram of sodium carbonate is heated in a platinum crucible for two hours over a small flame, weighed, and placed in an erlenmeyer together with twenty cubic centimeters of the sulfuric acid, care being taken to avoid loss from the vigorous evolution of carbon dioxid. After boiling for ten minutes all the carbon dioxid is removed from solution. After cooling, the excess of acid is determined by titration with a standardized barium hydroxid solution, using rosolic acid as indicator.
The solution of barium hydroxid is made as follows: Digest, with warm water, 260 grams of caustic baryta, Ba(OH)₂, until it is nearly all dissolved, filter, and make up to a volume of ten liters and keep in a flask free of carbon dioxid. A solution of barium hydroxid is to be preferred to the corresponding sodium compound for titration. If traces of carbonate be formed in the two liquids, the sodium salt will remain in solution while the barium compound will settle at the bottom of the flask. [Pg 204]
The Indicator.—The indicator used to determine the end of the reaction is made by dissolving one gram of rosolic acid in fifty cubic centimeters of alcohol. From one to two drops are enough for each titration. The color reaction is less definite as the quantity of ammonia in the liquid increases. When the titration solutions have been prepared as above described it is found to require about ninety of the barium hydroxid to neutralize twenty cubic centimeters of the sulfuric acid.
By direct titration with sodium carbonate it is ascertained how many grams of nitrogen the twenty cubic centimeters of sulfuric acid represent.
Example.—Suppose the weight of the dried sodium carbonate prepared as above directed is 0.6989 gram.
| ½Na₂CO₃, | ½N₂ | ||||
| Then 0.6989 | : | 53 | = x | : | 14 |
Whence x = 0.184615 gram of nitrogen.
Suppose further that twenty cubic centimeters of sulfuric acid solution require ninety-four cubic centimeters of barium hydroxid for complete saturation and after treatment with the above amount of sodium carbonate, ten and a half cubic centimeters of the barium solution to neutralize the remaining acid.
Then 94 - 10.5 = 83.5
And 0.184615: 83.5 = x: 94.
Whence x = 0.207830 gram of nitrogen corresponding to twenty cubic centimeters of the sulfuric acid used.
Then 0.20783 ÷ 94 = 0.002211 gram of nitrogen corresponding to one cubic centimeter of the barium hydroxid solution.
If then in the analysis of a fertilizer it is found that 60.5 cubic centimeters are required to neutralize the excess of sulfuric acid after distillation the percentage of nitrogen in the sample is found as follows:
60.5 × 0.002211 = 0.13377.
0.20783 - 0.13377 = 0.07406.
0.07406 × 100 = 7.406 = per cent nitrogen in sample when one gram is taken for the combustion.
184. The Official Kjeldahl Method. Not Applicable in the Presence of Nitrates.[153]—Reagents.—(1) Acids.—(a) Standard hydrochloric acid the [Pg 205] absolute strength of which has been determined by precipitating with silver nitrate, and weighing the silver chlorid as follows:
To any convenient quantity of the acid to be standardized, add solution of silver nitrate in slight excess, and two cubic centimeters of pure nitric acid, of specific gravity 1.2. Heat to boiling-point, and keep at this temperature for some minutes without allowing violent ebullition, constantly stirring until the precipitate assumes the granular form. Allow to cool somewhat, and then pass the fluid through the asbestos. Wash the precipitate by decantation, with 200 cubic centimeters of very hot water, to which have been added eight cubic centimeters of nitric acid and two cubic centimeters of dilute solution of silver nitrate containing one gram of the salt in 100 cubic centimeters of water. The washing by decantation is performed by adding the hot mixture in small quantities at a time, and beating up the precipitate well with a thin glass rod after each addition. The pump is kept in action all the time, but to keep out dust during the washing the cover is only removed from the crucible when the fluid is to be added.
Put the vessels containing the precipitate aside, return the washings once through the asbestos so as to obtain them quite clear, remove them from the receiver, and set aside to recover the excess of silver. Rinse the receiver and complete the washing of the precipitate with about 200 cubic centimeters of cold water. Half of this is used to wash by decantation and the remainder to transfer the precipitate to the crucible with the aid of a trimmed feather. Finish washing in the crucible, the lumps of silver chlorid being broken down with a glass rod. Remove the second filtrate from the receiver and pass about twenty cubic centimeters of ninety-eight per cent alcohol through the precipitate. Dry at from 140° to 150°. Exposure for half an hour is found more than sufficient, at this temperature, to dry the precipitate thoroughly.
(b) Standard sulfuric acid, the absolute strength of which has been determined by precipitation with barium chlorid and weighing the resulting barium sulfate:
For ordinary work half normal acid is recommended, i. e., containing 18.2285 grams of hydrochloric or 24.5185 grams sulfuric acid [Pg 206] to the liter; for work in determining very small amounts of nitrogen, one-tenth normal acid is recommended. In titrating mineral acids against ammonia solutions, use cochineal as indicator.
(c) Sulfuric acid, specific gravity 1.84, free of nitrates and also of ammonium sulfate, which is sometimes added in the process of manufacture to destroy nitrogen oxids:
(2) Standard alkali, the strength of which, relative to the acid, has been accurately determined. One-tenth normal ammonia solution, i. e., containing 1.7051 grams of ammonia to the liter, is recommended for accurate work:
(3) Metallic mercury or mercuric oxid, prepared in the wet way: That prepared from mercuric nitrate can not be safely used.
(4) Potassium permanganate finely pulverized:
(5) Granulated zinc, pumice stone, or one-half gram of zinc dust is to be added to the contents of the flasks in distillation, when found necessary, in order to prevent bumping:
(6) Potassium sulfid.—A solution of forty grams of commercial potassium sulfid in one liter of water:
(7) Soda.—A saturated solution of sodium hydroxid free of nitrates:
(8) Indicator.—Solution of cochineal prepared as follows: Tincture of cochineal is prepared by digesting for a day or two, at ordinary temperatures, and frequently agitating, three grams of pulverized cochineal in a mixture of fifty cubic centimeters of strong alcohol with 200 cubic centimeters of distilled water. The solution is decanted or filtered through Swedish paper.
Apparatus.—(1) Kjeldahl digestion flasks of hard, moderately thick, well-annealed glass: These flasks are about twenty-two centimeters long, with a round, pear-shaped bottom, having a maximum diameter of six centimeters and tapering out gradually in a long neck, which is two centimeters in diameter at the narrowest part, and flared a little at the edge. The total capacity is from 225 to 250 cubic centimeters.
(2) Distillation flasks of ordinary shape, of 550 cubic centimeters capacity, or preferably flasks of well-annealed glass, of the same capacity, of pear-shaped bottom, for both digestion and [Pg 207] distillation, fitted with a rubber stopper and a bulb-tube above to prevent the possibility of sodium hydroxid being carried over mechanically during distillation: The bulbs are about three centimeters in diameter, the tubes being of the same diameter as the condenser and cut off obliquely at the lower end. This is adjusted to the tube of the condenser by a rubber tube.
Manipulation.—(1) The Digestion.—From seven-tenths to three and five-tenths grams of the substance to be analyzed, according to its proportion of nitrogen, are brought into a digestion flask with approximately seven-tenths gram of mercuric oxid or its equivalent in metallic mercury and twenty cubic centimeters of sulfuric acid. The flask is placed in an inclined position, and heated below the boiling-point of the acid for from five to fifteen minutes or until frothing has ceased. If the mixture froth badly, a small piece of paraffin may be added to prevent it. The heat is then raised until the acid boils briskly. No further attention is required until the contents of the flask have become a clear liquid, which is colorless or at least has only a very pale straw color. The flask is then removed from the frame, held upright, and while still hot, potassium permanganate is dropped in carefully and in small quantities at a time until, after shaking, the liquid remains of a green or purple color.
(2) The distillation.—After cooling, the contents of the flask are transferred to the distilling flask with about 200 cubic centimeters of water, a few pieces of granulated zinc, pumice stone, or one-half gram of zinc dust when found necessary to keep the contents of the flask from bumping, and twenty-five cubic centimeters of potassium sulfid solution are added, shaking the flask to mix its contents. Next add fifty cubic centimeters of the soda solution, or sufficient to make the reaction strongly alkaline, pouring it down the sides of the flask so that it does not mix at once with the acid solution. Connect the flask with the condenser, mix the contents by shaking, and distil until all ammonia has passed over into the standard acid. The first 150 cubic centimeters of the distillate will generally contain all the ammonia. This operation usually requires from forty minutes to one hour and a half. The distillate is then titrated with standard alkali. [Pg 208]
The use of mercuric oxid in this operation greatly shortens the time necessary for digestion, which is rarely over an hour and a half in case of substances most difficult to oxidize, and is more commonly less than an hour. In most cases the use of potassium permanganate is quite unnecessary, but it is believed that in exceptional cases it is required for complete oxidation, and in view of the uncertainty it is always used. The potassium sulfid removes all the mercury from the solution, and so prevents the formation of mercurammonium compounds which are not completely decomposed by soda solution. The addition of zinc gives rise to an evolution of hydrogen and prevents violent bumping. Previous to use, the reagents should be tested by a blank experiment with sugar, which will partially reduce any nitrates that are present, which might otherwise escape notice.
Figure. 13.
Distilling Apparatus.
185. The Distillation Apparatus in Use in the Laboratory of the Department of Agriculture.—In this laboratory the distilling apparatus is arranged as shown in Figure 13. The flasks are the same as are used in the digestion. They are connected to the block tin condensers by the bulb and rubber tubes, shown hanging on the projecting ends of the block tin condensers on the right and left of [Pg 209] the figure. The condensers are contained in a trough through which cold water flows during the distillation. The bulb above the flask carries an emergent tube which extends to near the center of the bulb and is bent laterally to avoid any danger of carrying over any alkali that may be projected into the bulb during boiling. The boiling is continued usually for nearly an hour or until bumping begins. The table on which the apparatus is placed is so arranged as to permit of easy access on all sides. The standard acid is held in erlenmeyers placed on wooden blocks so that the end of the condenser which is a drawn-out glass tube, dips beneath the surface of the acid.
186. Patrick’s Distilling Flask.—To avoid the expense and annoyance attending the breaking of the distilling flasks Patrick has proposed to make them of copper.[154] The size, about half a liter, made for the evolution of oxygen for experimental purposes, may be used. A little excess of potassium sulfid is used to make up for any of it which might be consumed by the copper. About twenty-five cubic centimeters of this solution are recommended. No zinc or pumice stone is required to prevent bumping and the distillation may be finished within thirty minutes, thus securing a saving of time. There will doubtless be a slight corrosion of the flasks by the sulfid employed but where the gunning oxidation process is practiced this danger would be avoided.
187. Modifications of the Kjeldahl Process.—It would be impracticable here to give even a summary of the many unimportant changes which the moist combustion process has undergone since the first papers of its author were published. These changes may be divided into three classes; viz., 1. Those changes which refer solely to the quantities of substance taken for analysis, to the composition of the acid mixture, to the duration of the digestion, to the form and size of the flasks, both for digestion and distillation, and to the manner of distillation and of titration. For references to the papers on these subjects the reader may consult the periodic journals.[155] The most important of these minor changes are the following: Instead of the titration by means of separated iodin most chemists have had recourse to [Pg 210] the simpler method of direct titration of the excess of acid by a set solution of an alkali. Barium, sodium, and potassium hydroxids are the alkaline solutions most employed. This process permits of a larger quantity of the sample being taken for combustion and of the use of a larger quantity of acid in the receiver. It also implies the use of a larger digestion flask. In fact it is now quite common to make the digestion in a special glass flask large enough to be used also for the distillation. This saves one transfer of the material with the possible danger of loss attending it.
In the distillation it is a common practice, especially in Germany, to do away with the condensing worm and to carry a long glass tube from the distilling flask directly into the acid on the receiver. The only inconvenience in this method is the heating of the contents of the receiving flask, but this is attended with no danger of loss of ammonia and the distillate, on account of the high temperature it acquires, is left free of carbon dioxid. Many of these minor changes have tended to simplify the process, but without affecting the principle of the method in the least.
2. In the second place a class of changes may be mentioned in which there is a marked difference in the method of effecting the oxidation secured by the introduction of a substance, usually a metal, during the digestion for the purpose of accelerating the oxidation. In the original process the only aid to oxidation was applied at the end of the digestion in the use of potassium permanganate. In the modifications now under consideration a metallic oxid or metal is applied at the beginning of the digestion. Copper and mercury are the metals usually employed. A separate paragraph will be given to the description of this modification known as the process of Wilfarth.
3. The third class of changes is even more radical in its nature, having for its object the adaptation of the moist combustion method to oxidized or mineral nitrogen. The chief feature of this class of changes consists in the introduction of a substance rich in hydrocarbons, and capable of easily forming nitro compounds, for the purpose of holding the oxids of nitrogen which are formed during the combustion and helping finally to reduce them to the form of ammonia. [Pg 211] The chief varieties of this class of changes were proposed by Asboth, Jodlbaur, and Scovell, and will be fully set forth in separate paragraphs.
188. Method of Wilfarth.—The basis of this modification as already noted rests on the fact that certain metallic oxids have the power of carrying oxygen, and thus assisting in a katalytic way in the combustion of organic matter.[156] The copper and mercury oxids are best adapted for this purpose and experience has shown that mercuric oxid, or even metallic mercury gives the best results. The manipulation is carried out as follows: From one to three grams of the sample, according to its richness in nitrogen, are heated with a mixture of twenty cubic centimeters of acid containing two-fifths fuming and three-fifths ordinary sulfuric acid. To this is added about seven-tenths gram of mercuric oxid prepared in the wet way from a mercury salt free of nitrogen. The combustion takes place in the usual kjeldahl flask. If the boiling be continued until the liquid is entirely colorless, final oxidation with potassium permanganate is unnecessary. To save time the combustion may be stopped when a light amber color is reached, and then the oxidation finished with permanganate. Before distilling, a sufficient quantity of potassium sulfid is added to precipitate all the mercury as sulfid and thus prevent the formation of mercurammonium compounds which would produce a deficit of ammonia. A convenient strength of the sulfid solution is obtained by dissolving forty grams of potassium sulfid in one liter of water. Bumping at the end of the distillation is not usual, especially if potash-lye be used, but should it occur it may be stopped by the addition of zinc dust.
Only when a large excess of potassium sulfid is used is there an evolution of hydrogen sulfid, the presence of which, however, does not influence the accuracy of the results.
The presence of mercuric sulfid in the solution tends to prevent bumping during the distillation, but it is advisable, nevertheless, to use a little zinc dust. Other minor modifications consist of forming the acid mixture with equal volumes of concentrated and fuming sulfuric acid containing in one liter 100 grams of phosphoric acid anhydrid,[157] and using metallic mercury instead of mercuric oxid; or a mixture of half a gram of copper sulfate and one gram of metallic mercury,[158] or half a gram of copper oxid and a few drops of platinic chlorid solution [Pg 212] containing 0.04 gram of platinum in a cubic centimeter.[159]
189. Modification of Asboth.—In order to adapt the moist combustion process to nitric nitrogen Asboth proposed the use of benzoic acid.[160] For half a gram of saltpeter 1.75 grams of benzoic acid should be used. At the end of the combustion the residual benzoic acid is oxidized by means of potassium permanganate with a subsequent reheating. If the nitrogen be present as an oxid or as cyanid, one gram of sugar is added. The metallic element added is half a gram of copper oxid. Asboth also recommends that the soda-lye used in the distillation be mixed with sodium potassium tartrate for the purpose of holding the copper and manganese oxids in solution and thus preventing bumping. The alkaline liquor contains in one liter 350 grams of the double tartrate and 300 grams of sodium hydroxid.
The principle on which the use of benzoic acid rests is found in the fact that it easily yields nitro-compounds and thus prevents the loss of the nitrogen oxids, these readily combining with the benzoic acid. The nitro-compounds can be subsequently converted into ammonia by treatment with potassium permanganate.
The pyridin and chinolin groups of bodies do not yield all their nitrogen as ammonia by the above treatment.
The conclusions drawn by Asboth from the analytical data obtained were:
(1) Sugar should be used in the ordinary kjeldahl process in those cases where the nitrogen in the organic substance is present as oxids or as cyanogen.
(2) In the case of nitrates good results may be secured with benzoic acid but permanganate must be added at the end.
(3) The kjeldahl-wilfarth process can be applied with substances difficultly decomposed, e. g., alkaloidal bodies.
190. Variation of Jodlbaur.—The benzoic acid method, although a step forward, is not entirely satisfactory in the treatment of nitrates by moist combustion. Jodlbaur has proposed to substitute for the benzoic, phenolsulfuric acid.[161]
From two to five-tenths gram of a nitrate are treated with twenty cubic centimeters of concentrated sulfuric and two and a half of [Pg 213] phenolsulfuric acid, together with three grams of zinc dust and five drops of a solution of platinic chlorid of the strength mentioned above. The phenolsulfuric acid is prepared by dissolving fifty grams of phenol in 100 cubic centimeters of strong sulfuric acid. The combustion is continued until the solution is colorless, which may take as much as five hours. If phosphoric acid anhydrid be used as recommended above, the time of the combustion may be diminished by one-half, but in such a case the glass of the combustion flask is strongly attacked and is quite likely to break.
With substances very rich in nitrates it is advisable to rub them first with dry gypsum.
The theory of the process rests on the fact that by a careful admixture of a nitrogenous substance diluted with land plaster, with phenolsulfuric acid, it is possible to change the nitric acid into nitro-phenol, and by the reducing action of zinc dust to change the nitro-product formed into amido-phenol. This afterwards is transformed into ammonium sulfate by heating with sulfuric acid, by which process, at the same time, all other nitrogenous compounds present in the substance, as with Kjeldahl’s method, likewise form ammonium sulfate, only with the difference that addition of mercury is here absolutely necessary for the complete transformation of the slowly decomposed amido-phenol which again brings about the necessity of decomposing the nitrogenous mercury compounds formed in the solution by potassium sulfid, which is added after or with the soda-lye.
191. The Dutch Jodlbaur Method.—The Royal Experiment Station of Holland directs that the jodlbaur process be carried out as indicated below.[162]
The reagents necessary are:
1. Phenolsulfuric acid, prepared by dissolving 100 grams of pure crystallized phenol in pure sulfuric acid (1.84) and making up the solution to a liter with the same sulfuric acid:
2. Zinc, carefully washed and thoroughly dried:
3. Sodium hydroxid solution, the same as is used in the kjeldahl method:
4. Potassium sulfid solution, made by dissolving 355 grams of potassium sulfid (K₂S), or sodium sulfid solution, made by dissolving 250 grams of sodium sulfid (Na₂S) in a liter of water. [Pg 214]
As apparatus, are necessary oxidation flasks holding about 200 cubic centimeters, and distillation flasks holding about 750 cubic centimeters, both of bohemian glass.
Manipulation.—Weigh one gram of substance, moisten it with water, dry, and introduce into an oxidation flask. Cover with fifteen cubic centimeters of phenolsulfuric acid and, after cooling, thoroughly mix by gently shaking. After five minutes add from two to three grams of zinc in small proportions, keeping the flask cool, then twenty cubic centimeters of sulfuric acid, and finally two drops of mercury. Boil the mixture till the fluid is colorless. Cool and dilute. Wash into a distillation flask and add an excess of sodium hydroxid solution and twenty-five cubic centimeters of the sodium (or potassium) sulfid solution. Distil and titrate as in the kjeldahl method.
192. The Halle-Jodlbaur Method.—At the Halle station it is the uniform practice to mix the nitrate with gypsum before the combustion.[163] In the case of Chile phosphates ten grams are rubbed with an equal amount of gypsum, and two grams of the mixture, equal to one gram of the nitrate, taken for the determination. In the case of saltpeter mixtures which contain over eight per cent of nitrogen, one gram of the mixture with gypsum is taken, of guanos one and a half grams, and of lower forms of nitrates or mixtures thereof, from three to five grams.
The sample, as prepared above, is treated with thirty cubic centimeters of a mixture of phenolsulfuric acid and phosphoric acid anhydrid. The mixture is prepared by dissolving sixty-six grams of phenol and 250 grams of phosphoric anhydrid in strong sulfuric acid, and, after cooling, mixing the two solutions and making the volume up to 1,650 cubic centimeters with pure sulfuric acid. The mixture contains, in thirty cubic centimeters, one and two-tenths grams of phenol and four grams of phosphoric anhydrid. In the use of phenolsulfuric acid the the presence of phosphoric anhydrid is indispensable in keeping the sulfuric acid water-free and in absorbing the water produced by the combustion.
The phenolsulfuric acid used contains only enough phenol to reduce half a gram of saltpeter.
The sample and acid mixture having been put in the combustion flask the [Pg 215] latter is shaken, at intervals, for an hour, and the contents cooled.
The conversion of the nitrates into nitro-phenol compounds is finished in this time, and the next step consists in reducing these bodies to the amido-phenol group. This is accomplished in the cold by nascent hydrogen produced by the addition of zinc dust to the mixture. From one to three grams of the dust are to be used in proportion to the quantity of nitrates originally present.
The flask should be placed in a cooling mixture and the zinc dust added in small portions to prevent a too violent evolution of hydrogen. After the reduction is ended the flask is allowed to stand for two hours, after which the combustion, distillation, and titration are accomplished in the usual way. On cooling, after the end of the combustion, the contents of the flask become solid. They may be brought again into liquid state by shaking and gentle warming.
193. The Official Kjeldahl Method for Nitric Nitrogen.—As has already been stated, the presence of certain organic compounds, rich in hydrocarbons, permits the reduction of nitric nitrogen to ammonia by combustion with sulfuric acid. Benzol, phenol, and salicylic acid have all been used for this purpose. The official chemists have adopted for their method the salicylic acid process first proposed by Scovell.[164]
Besides the reagents and apparatus given under the Kjeldahl method there will be needed:
(1) Zinc dust: This should be an impalpable powder; granulated zinc or zinc filings will not answer.
(2) Sodium thiosulfate:
(3) Commercial salicylic acid:
It is found most convenient to prepare a solution of 33.3 grams of salicylic acid in one liter of the strongest sulfuric acid, and keep it for use rather than to mix it for each combustion. We prefer the thiosulfate process first mentioned below. In the zinc dust method there has been noticed a tendency for the distillation flask to break just at the end of the process.
The Manipulation.—Place from seven-tenths to three and five-tenths grams of the substance to be analyzed in a kjeldahl digesting flask, add sixty cubic centimeters of sulfuric acid containing [Pg 216] one gram of salicylic acid, and shake until thoroughly mixed, then add five grams of crystallized sodium thiosulfate; or add to the substance thirty cubic centimeters of sulfuric acid containing two grams of salicylic acid, then add gradually two grams of zinc dust, shaking the contents of the flask at the same time. Finally place the flask on the stand for holding the digestion flasks, where it is heated over a low flame until all danger from frothing has passed. The heat is then raised until the acid boils briskly and the boiling continued until white fumes no longer pour out of the flask. This requires about five or ten minutes. Add now approximately seven-tenths gram of mercuric oxid or its equivalent in metallic mercury, and continue the boiling until the liquid in the flask is colorless or nearly so. In case the contents of the flask are likely to become solid before this point is reached add ten cubic centimeters more of sulfuric acid. Complete the oxidation with a little potassium permanganate in the usual way, and proceed with the distillation as described in the kjeldahl method. The reagents should be tested by blank experiments.
194. The Gunning Moist Combustion Process.—The modification proposed by Gunning was based upon the observation that in the ordinary kjeldahl process the excess of sulfur trioxid in the beginning of the operation soon escapes or unites with water in a form not easily decomposed.[165] During the progress of the combustion the acid diminishes in strength until it is below the concentration represented by the formula H₂SO₄, and in this diluted condition the oxidation takes place more slowly. Gunning proposes to avoid this difficulty by mixing potassium sulfate with the sulfuric acid. This salt forms with the sulfuric acid, acid salts which, by heating, lose water easier than acid and as is well known, they not only act as decomposing and oxidizing media as well as sulfuric acid, but even in a higher degree, resembling the action of sulfuric acid at high temperatures and under pressure.
By heating this mixture of sulfuric acid and potassium sulfate with organic matters in an open vessel, not only the water originally present, but that which is formed during the oxidation is driven off without loss of acid. For this reason instead of the oxidizing mixture [Pg 217] becoming weaker, the acid becomes stronger, the boiling-point rises and this, combined with the fluidity of the mass favors the decomposition and oxidation of the organic matter in a constantly increasing ratio.
The original mixture used by Gunning had the following composition; viz., one part of potassium sulfate and two parts of strong sulfuric acid. The substances are united by heat and, on cooling, are in a semi-solid state, melting, however, easily on the application of heat and assuming a condition to be easily poured from vessel to vessel. The quantity of the sample taken should vary in proportion to its nitrogenous content from half a gram to a gram. The combustion takes place in flasks entirely similar to those used in the ordinary kjeldahl process. In the case of liquids, they should be previously evaporated to dryness before the addition of the oxidizing mixture. At the beginning of the combustion there is a violent foaming attended with evolution of some acid and much water, and afterwards of stronger acid. This loss of acid should not be allowed to go far enough to produce too great concentration of the material in the flask. One of the best ways to avoid it is to place a funnel in the flask covered with a watch-glass which will permit of the condensation and return of the escaping acid. As soon as the foaming ceases, the flame should be so regulated as to permit of the volatilized acid being condensed upon the sides of the flask. In the end a colorless mass is obtained in which no metallic oxids are present, and this mass can at once be diluted with water, treated with alkali, and distilled. According to the nature of the substance from half an hour to an hour and a half are required for the complete combustion.
Modifications of the Gunning Method.—As in the case of the kjeldahl method, numerous minor modifications of the gunning method have been made, the most important of which relate to its application to substances containing nitrates. In general the same processes are employed in this case as with the kjeldahl method. One of the best modifications consists in the use of the mixture of salicylic and sulfuric acids followed by the addition of sodium thiosulfate or of potassium sulfate or sulfid. These modifications will be given in detail under the official methods. [Pg 218]
195. Reactions of the Gunning Process.—The various reactions which take place during the combustion according to the gunning method have been tabulated by Van Slyke.[166]
The first reaction to take place is the union of sulfuric acid and potassium sulfate to form potassium acid sulfate in accordance with the following equation:
(1) K₂SO₄ + H₂SO₄ = 2KHSO₄.
When heated, the potassium acid sulfate decomposes, forming potassium disulfate and water, thus:
(2) 2KHSO₄ = K₂S₂O₇ + H₂O.
The potassium disulfate at a higher temperature decomposes, forming normal potassium sulfate and sulfur trioxid, thus:
(3) K₂S₂O₇ = K₂SO₄ + SO₃.
At a sufficiently high temperature the two preceding reactions may take place in one, thus:
2KHSO₄ = K₂SO₄ + H₂O + SO₃.
At the temperature at which these reactions take place, the water that is set free does not recombine with the sulfur trioxid nor with the sulfuric acid that is present in excess, but is expelled from the mixture; hence the mixture becomes more concentrated during the digestion. The sulfur trioxid set free acts upon the organic matter in the powerful manner peculiar to it, and the potassium sulfate formed in the last reaction above unites with another molecule of sulfuric acid, and the same round of reactions is repeated continuously so long as there is an excess of free sulfuric acid present in the mixture. As the liquid becomes more concentrated with the continuation of the digestion, the boiling-point increases so that the effect is the same as heating under pressure. The danger of too great concentration and risk of consequent loss of nitrogen is avoided by using increased proportions of sulfuric acid.
As compared with the kjeldahl the gunning method presents the following advantages:
(1) The gunning method requires fewer reagents. As no form of mercury is used no potassium sulfid is needed, and there is no risk of loss from the presence of mercurammonium compounds.
(2) The solution to which caustic soda is added is clear, so that in [Pg 219] neutralizing, it is an easy matter to avoid great excess of alkali, and so, in most cases, to avoid foaming and bumping in distillation.
(3) In the blank determinations less nitrogen is found in the reagents used in the gunning method. In only one case was more nitrogen reported in a blank by this method; in all the others the amount averaged considerably less.
196. The Official Gunning Method.—In a digestion flask holding from 250 to 500 cubic centimeters place from seven-tenths to two and eight-tenths grams of the substance to be analyzed, according to its proportion of nitrogen. Then add ten grams of powdered potassium sulfate and from fifteen to twenty-five cubic centimeters (ordinarily about twenty cubic centimeters) of concentrated sulfuric acid. Conduct the digestion as in the kjeldahl process, starting with a temperature below boiling-point and increasing the heat gradually until frothing ceases. Digest until colorless or nearly so. Do not add either potassium permanganate or potassium sulfid. Dilute, neutralize, and distil as in the kjeldahl method. In neutralizing, it is convenient to add a few drops of phenolphthalein indicator, by which one can tell when the acid is completely neutralized, remembering that the pink color, which indicates an alkaline reaction, is destroyed by a considerable excess of strong fixed alkali. The distillation and titration are conducted as in the kjeldahl method. In distilling, the use of zinc or of any substance to prevent bumping or foaming is generally unnecessary, if too great an excess of fixed alkali be avoided. The amount of sulfuric acid recommended by Gunning is two grams for each gram of potassium sulfate; but Voorhees has found that this mixture is so viscous as to cause troublesome foaming frequently, and after cooling it cakes in a hard mass, which may be difficult to redissolve.[167] To avoid foaming and caking, he has found it an effective means to increase the amount of sulfuric acid used, taking instead of two grams to one of potassium sulfate three or four grams of acid to one of potassium sulfate. It is, therefore, suggested in carrying out the work, to use from five to twenty-five cubic centimeters (ordinarily about twenty cubic centimeters) of sulfuric acid for ten grams of [Pg 220] potassium sulfate. In case the potassium sulfate is not free from nitrogen compounds, one or two recrystallizations will make it pure.
197. Gunning Method Adapted to Nitrates.—The essential features of this modification are due to Winton and Voorhees.[168] The modifications of the kjeldahl method, for similar purposes, furnished the material details for the gunning modified process. Winton reports good results from digesting for two hours, from half a gram to a gram of the sample with thirty cubic centimeters of sulfuric containing two grams of salicylic acid, in a flask of half a liter capacity. Two grams of zinc dust are then slowly added, with constant shaking, and the flask heated, at first gently, until, after a few minutes boiling, dense fumes are no longer emitted. Three grams of potassium sulfate are next added and the boiling continued until the solution is colorless, or if iron be present, until a light straw color is produced. On cooling, when the mixture begins to solidify, water is added with caution, and afterwards sodium hydroxid, and the ammonia is obtained by distillation.
In the process, as conducted by Voorhees, about one gram of the sample is digested with ten grams of potassium sulfate and thirty cubic centimeters of sulfuric containing one gram of salicylic acid, and three grains of zinc sulfid. The heat is kept down until frothing ceases, and then the mass kept in gentle ebullition until clear. The distillation is accomplished with the usual precautions. The voorhees process is superior to that recommended by Winton in adding the potassium sulfate at the beginning of the combustion.
198. Official Gunning Method Modified to Include the Nitrogen of Nitrates.—In a digestion flask holding from 250 to 500 cubic centimeters, place from seven-tenths to three and five-tenths grams of the substance to be analyzed, according to the amount of nitrogen present. Add from thirty to thirty-five cubic centimeters of salicylic acid mixture, namely, thirty cubic centimeters of sulfuric to one gram of salicylic acid, shake until thoroughly mixed, and allow to stand from five to ten minutes, with frequent shaking; then add five grams of sodium thiosulfate and ten grams of potassium sulfate. Heat very gently until frothing ceases, then strongly until nearly colorless. Dilute, neutralize, and distil the same as in the gunning method. [Pg 221]
199. Introductory Considerations.—In the foregoing pages has been given a summary of the methods most in vogue for the estimation of nitrogen in fertilizers and fertilizing materials. There are many cases in which the analyst may have to deal with a definite chemical compound, and where a modified or shorter method may be used. There are other cases in which the nitrogen may be present in two or three definite forms, as in artificially mixed fertilizers, and where it is desirable to show the proportions in which the various forms are present. For these reasons it is necessary to be able to use methods by which the percentage of nitrogen in its various forms may be relatively as well as absolutely determined. Such a case would be presented for instance, in that of a fertilizer containing dried blood, sodium nitrate, and ammonium sulfate. It is evident here that the total nitrogen could be determined by the volumetric method by combustion with copper oxid, or by the moist combustion process adapted to nitric nitrogen, but the method of determining the percentage of each constituent has not yet been described.
We have to deal here with a case entirely similar to that of phosphoric acid in a superphosphate. There is no doubt whatever of the uneven assimilability of the different forms of nitrogen. A nitrate, for instance, is already in condition for assimilation by plants. An ammoniacal salt is only partly changed to a state suited to plant nutrition while organic nitrogen is forced to undergo a complete transformation before it becomes available to supply the needs of the growing plant. It is important, therefore, equally to the analyst, the merchant, and the agronomist, to know definitely the forms of combination in which the nitrogen exists and the relative proportion of the different combinations.
200. Nitrogen as Ammonia.—The most frequent form in which nitrogen as ammonia is used for fertilizing is as sulfate. The method of determination to be described is, however, equally applicable to all ammonia salts. When no other form of nitrogenous compound is present the ammonia can be easily and directly determined by distillation with soda- or potash-lye, as described in the final part of the moist combustion process. [Pg 222]
To one gram of the ammonia salt add from 200 to 300 cubic centimeters of water and thirty grams of the soda-lye used in the moist combustion process; distil, collect the ammonia, and titrate the excess of sulfuric acid exactly as there described.
Fresenius recommends that the ammonia expelled by distillation be taken up by one-fifth normal sulfuric acid, the excess of which is titrated with one-fifth normal soda, using phenolphthalein as an indicator. If the distillate, on examination, be found to contain thiocyanate, soda-lye cannot be used for the expulsion of ammonia, but, in its place, caustic magnesia is applied.
In all cases where organic matter containing nitrogen is present, caustic magnesia must be substituted for the soda solution. The magnesia must be added in sufficient excess and the distillation continued a little longer than is necessary when soda-lye is used. Otherwise the details of the operation are the same.
In a mixed fertilizer containing organic nitrogen and ammonia salts, the total nitrogen can be determined by the moist combustion process, and the ammoniacal nitrogen by distillation with magnesia. The difference between the two results will give the nitrogen due to the organic matter.
To avoid any danger whatever of decomposing organic nitrogenous compounds, the ammonia may be determined in the cold by treatment with soda-lye, under a bell-jar containing some set sulfuric acid. The operation must be allowed to continue for many days. Even at the end of a long time it will be found that some ammonia is still escaping. It may therefore be finally inferred that all the nitrogen as ammonia is not obtained by this process, or that even magnesia may gradually convert other nitrogenous compounds into ammonia. In this connection the methods of determining ammonia in soils 406, 407, and 408 of volume one may be consulted.
201. Method of Boussingault.—The official French method is essentially the original method of Boussingault with slight modifications. It is conducted as follows:[169] In case the sample is ammonium sulfate about half a gram is placed in a flask of half a liter capacity, together with 300 cubic centimeters of distilled water and two grams of caustic magnesia. The flask is connected with a condenser [Pg 223] of glass or metal which ends in a tube drawn out to a point and dipping beneath the set acid in the receiver in the usual way. The acid is colored with litmus or lacmoid tincture. The distillation is continued until about 100 cubic centimeters have gone over. The receiver is then removed with the usual precautions and the residual acid titrated. Suppose twenty cubic centimeters of normal acid have been employed and twelve and a half cubic centimeters of normal alkali be necessary to neutralize the excess of the acid. Then the nitrogen is found by the following equations: 20.0 - 12.5 = 7.5 and 7.5 × 0.014 = 0.105 gram = weight of nitrogen found. Then 0.105 × 100 ÷ 5 = 21.00 = per cent of nitrogen found.
The distilling apparatus of Aubin is preferred by the French chemists, an apparatus so arranged with a reflux partial condenser, that nearly all the aqueous vapor is returned in a condensed state to the flask while the ammonia, on account of its great volatility, is carried over into the receiver. To avoid the regurgitation which might be caused by the concentrated ammonia gas coming in contact with the acid the separable part of the condensing tube is expanded into a bulb large enough to hold all the acid which lies above its mouth. By the means of this apparatus the ammonia is all collected in the standard acid without greatly increasing its volume and the titration is thus rendered sharper. The employment of caustic magnesia has the advantage of not decomposing any organic matters or cyanids that may be present.
If the sample under examination hold part of its ammonia as ammonium magnesium phosphate it will be necessary first to treat it with sulfuric acid in order to set the ammonia free and then to use enough of the magnesium oxid to neutralize the excess of the sulfuric acid and still supply the two grams necessary for the distillation. When the sample contains a considerable quantity of organic matter it sometimes tends to become frothy towards the end of the distillation. This trouble can be avoided by introducing into the flask one or two grams of paraffin.
Where carbon dioxid is given off during the distillation the contents of the receiver must be boiled before titration, or else lacmoid must be used as an indicator instead of litmus. [Pg 224]
202. Determination of Thiocyanates in Ammoniacal Fertilizers.—The extended use of ammonium sulfate as a fertilizer renders it important to determine the actual constituents which may be present in samples of this material. The following bodies have been found in commercial ammonium sulfates: Sulfuric acid, chlorin, ammonia, thiocyanic acid, potash, soda, lime and iron oxid. These are found in the soluble portions. In the insoluble portions have been found silica, sulfuric acid, lime, magnesia and iron oxid. A sample of commercial ammonium sulfate analyzed by Jumeau contained the following substances:[170]
| Per cent. | |
| Moisture | 10.5109 |
| Ammonium sulfate | 67.8453 |
| Ammonium thiocyanate | 9.3935 |
| Sodium sulfate | 9.2429 |
| Potassium sulfate | 0.9774 |
| Calcium sulfate | 0.6800 |
| Iron thiocyanate | 0.5000 |
| Magnesium chlorid | traces |
| Silica | 0.0830 |
| Undetermined | 0.7670 |
The determination of the thiocyanic acid in the thiocyanate is generally made by the oxidation of the sulfur to sulfuric acid and its subsequent weighing in the form of barium sulfate. Jumeau has modified the method by determining the amount of the thiocyanate by means of a titrated liquid. The method is practiced as follows:
A solution of ammonium thiocyanate is prepared, containing eight grams of this salt per liter, and its exact content of thiocyanate is rigorously determined by titration with silver nitrate or by the weight of the barium sulfate produced after the oxidation of the sulfur. Ten cubic centimeters of the titrated liquor are taken and diluted with water to about 100 cubic centimeters and ten cubic centimeters of pure sulfuric acid added. Afterward, drop by drop, a solution of potassium permanganate is added, containing about ten grams of that salt per liter. The permanganate is instantly decolorized. There is an evolution of hydrocyanic acid as the thiocyanate passes to the state of sulfuric acid. A single drop in excess gives to the mixture the well-known rose [Pg 225] coloration of the permanganate solution which persists for several hours. The number of cubic centimeters necessary to produce the persistent rose tint is noted and the same operation is carried on with from one-half to one gram of the unknown product which is to be assayed. A simple proportion indicates the content of the thiocyanate in the unknown body. The process is of great exactitude and permits the rapid determination of thiocyanic acid in the presence of chlorids, cyanids, etc., which remain without action upon the permanganate. In case chlorids and cyanids are absent the thiocyanate can be determined directly by silver nitrate either by weighing the precipitate or by the process of Volhardt based upon the precipitation of the silver by thiocyanate in the presence of a ferric salt. The end of the reaction is indicated by the red coloration which the liquid shows when the thiocyanate is in excess.
203. Separation of Albuminoid from Amid and Other Forms of Nitrogen in Organic Fertilizers.—It may be of interest to the dealer, farmer, and analyst, to discriminate between the albuminoid and other nitrogen in fertilizers, such as oil-cakes. The final value of the nitrogen for plant nourishment is not greatly different, but the immediate availability for nitrification is a matter of some importance. The most convenient process in such a case is the copper hydroxid separation process as improved by Stutzer.[171] The process is conveniently carried out in accordance with the method prescribed by the official chemists.[172]
Total Crude Protein.—Determine nitrogen as directed for nitrogen in fertilizers and multiply the result by 6.25 for the crude protein.
Determination of Albuminoid Nitrogen.—To from seven-tenths to eight-tenths gram of the substance in a beaker add 100 cubic centimeters of water, heat to boiling, or in the case of substances rich in starch, heat on the water-bath ten minutes, and add a quantity of cupric hydroxid mixture containing from one-half to six-tenths gram of the hydroxid; stir thoroughly, filter when cold, wash with cold water, and put the filter and its contents into the concentrated sulfuric acid for the determination of nitrogen. The filter-papers used must be practically free of nitrogen. Add sufficient potassium sulfid [Pg 226] solution to completely precipitate all copper and mercury, and proceed as in the moist combustion process for nitrogen. If the substance examined consist of seed of any kind, or residues of seeds, such as oil-cake or anything else rich in alkaline phosphates, add a few cubic centimeters of a concentrated solution of alum just before adding the cupric hydroxid, and mix well by stirring. This serves to decompose the alkaline phosphates. If this be not done cupric phosphate and free alkali may be formed, and the protein-copper may be partially dissolved in the alkaline liquid.
Cupric Hydroxid.—Prepare the cupric hydroxid as follows: Dissolve 100 grams of pure cupric sulfate in five liters of water, and add twenty-five cubic centimeters of glycerol; add a dilute solution of sodium hydroxid until the liquid is alkaline; filter, rub the precipitate up with water containing five cubic centimeters of glycerol per liter, and then wash by decantation or filtration until the washings are no longer alkaline. Rub the precipitate up again in a mortar with water containing ten per cent of glycerol, thus preparing a uniform gelatinous mass that can be measured out with a pipette. Determine the quantity of cupric hydroxid per cubic centimeter of this mixture.
Amid Nitrogen.—The albuminoid nitrogen determined as above subtracted from the total, gives that part of the organic nitrogen existing in the sample as amids and in other allied forms.
204. Separation of Nitric and Ammoniacal from Organic Nitrogen.—The nitrogen being present in three forms, viz., organic, ammoniacal, and nitric, the separation of the latter two may be accomplished by the following procedure:[173] One gram of the fertilizer is exhausted on a small filter with a two per cent solution of tannin, using from thirty to forty cubic centimeters in small portions. This is sufficient to dissolve all the nitrates and the greater portion of the ammoniacal salts, while the tannin renders insoluble all the organic nitrogenous compounds. The filter and its contents are treated for nitrogen by the kjeldahl process. When the distillation and titration are completed the solution obtained by the aqueous tannin is added to the distilling flask and the operation continued. This represents the ammoniacal nitrogen.
The nitric acid is estimated by the ferrous iron or other appropriate method in another portion of the substance. [Pg 227]
This method can be used even when the fertilizer contains ammonium magnesium phosphate. In this case digest one gram for fifteen hours in dilute soda-lye solution, which easily dissolves the ammonium magnesium phosphate. Filter and wash the insoluble part with the tannin solution. The residue is treated as above. The filtered solution distilled with soda-lye furnishes the ammonia. The nitrates are estimated by one of the methods above mentioned.
205. Nitric Nitrogen.—The methods of estimating nitric nitrogen, both when present in weighable quantities and as mere traces have been sufficiently described in the first volume. For convenience, however, the standard methods of procedure will be given here. The moist combustion methods adapted to nitrates and the volumetric copper oxid process have already been described. Of the reduction methods the process of Ulsch is one of the easiest of application and also reliable. As practiced by the official chemists the manipulation is conducted as described in the first volume, page 539.
206. Ulsch Method, Applicable to Mixed Fertilizers.—The method of Ulsch which is found to give good results with pure nitrates or with nitrates in the absence of other forms of nitrogen may also be adapted to mixed fertilizers containing nitrogen in more than one form. Street has developed such a method and shown, by analytical data, that it is applicable in a great number of cases.[174] The process is based on the substitution of magnesia for soda in the distillation and is carried on as follows:
Place one gram of the sample in a half liter flat-bottomed flask. Add about thirty cubic centimeters of water, one gram of reduced iron, and ten cubic centimeters of sulfuric acid diluted with an equal volume of water, shake well, and allow to stand for a short time. This will remove the danger of an explosion caused by the otherwise violent action which takes place. Close the neck of the flask with a rubber stopper through which passes a glass dropping-bulb filled with water. The flask having been stoppered, place it on a slab to which a moderate heat is applied. Allow the solution to come slowly to a boil and then boil for five minutes and cool. Add about 100 cubic centimeters of [Pg 228] water, a little paraffin, and about five grams of magnesium oxid. Boil for forty minutes, after which time all the ammonia will be distilled, and collect the ammonia in set acid.
The magnesia causes a slight frothing, which can easily be controlled by adding a little paraffin and by bringing to a boil very gradually. Fully forty minutes are necessary to distil all the ammonia. Tests were made after thirty minutes boiling and traces of ammonia were still found; after forty minutes these traces entirely disappeared.
The method is a quick one. One man can easily do six determinations at a time, and these six determinations can be made in but a little over an hour. Magnesia gives results closely agreeing with theory and causes a very slight frothing, which can be easily controlled. One gram of reduced iron is sufficient in all ordinary complete fertilizers.
Magnesia is preferred to caustic soda in the distillation because it produces less frothing and by reason of the danger of some of the soda-lye being carried over mechanically and thus tending to produce an error of a plus nature. In the use of magnesia, assurance must be had that it is strongly in excess. Being less active in its effects a longer time for the distillation must be taken than when soda-lye is used. The modified ulsch method just described is recommended provisionally and with the expectation that each analyst will ascertain its true merits before allowing it to displace longer approved processes.
207. Method of Schlösing-Wagner.—The Schlösing-Wagner method for estimating nitrogen in the nitrates of fertilizers is carried out at the Halle Experiment Station as follows:[175]
A flask, figure 14, of about 250 cubic centimeters capacity, is provided with a rubber stopper with two holes. Through one of them is passed the stem of a funnel carrying a glass stop-cock. The other carries a delivery-tube leading to the receiving vessel. The end of the delivery tube is bent so as to pass easily under the mouth of the measuring burette and is covered with a piece of rubber tubing.
Fifty cubic centimeters of saturated ferrous chlorid solution and the same quantity of ten per cent hydrochloric acid are placed in the flask. [Pg 229] The ferrous chlorid solution is obtained by dissolving nails or other small pieces of iron in hot hydrochloric acid and it is kept in glass stoppered flasks, of about fifty cubic centimeters capacity, entirely filled. The content of one flask is enough for about twelve determinations and by using the whole content of a flask as soon as possible after opening, any danger of oxidation which would take place in a large flask frequently opened is avoided.
Figure. 14.
Schlösing-Wagner Apparatus.
The contents of the flask are boiled until all the air is driven off. The delivery-tube is then placed under the measuring-tube, which is filled with forty per cent potash-lye. The measuring-tube is previously almost filled with potash-lye and then a few drops of water added and the tube covered with a piece of filter-paper. By a careful and quick inversion the measuring-tube can be brought into the vessel receiving it without any danger of air entering. The boiling is continued for some time and when no more air escapes, the end of the delivery-tube is brought into another freshly filled measuring-tube and the estimation is commenced.
Ten cubic centimeters of a normal saltpeter solution, containing two and a half grams of pure sodium nitrate in 100 cubic centimeters are placed in the funnel and, with continued boiling, allowed to pass, drop by drop, into the flask. When almost all has run out the funnel is [Pg 230] washed three times with ten cubic centimeters of ten per cent hydrochloric acid and this is allowed to pass, drop by drop, into the flask. When no more nitric oxid is evolved the measuring-tube is transferred to a large jar filled with distilled water.
The solution of the substance to be examined should be taken in such quantity as will give about the same quantity of gas as is furnished by the ten cubic centimeters test nitrate solution before described; viz., about seventy cubic centimeters. Eight or ten determinations can be made, one following the other, and at the end another determination with normal sodium nitrate solution should be made as a check. At the end of the operation all of the measuring-tubes are in the large jar filled with distilled water. The temperature of the surrounding water will soon be imparted to the contents of each tube and the volume of nitric oxid is read by bringing the level within and without the measuring-tube to the same point. The percentages are calculated for the given temperature and barometer pressure in the usual way; or to avoid computation the volume can be compared directly with the volume furnished by a normal nitrate solution, which is a much simpler method.
208. Schmitt’s Modified Method.—The method is a modification of that already described by the author in which a mixture of powdered zinc and iron is used as a reducing agent.[176] The process is carried out as follows: Ten grams of the nitrate are dissolved and the volume made up to half a liter. Ten cubic centimeters of glacial acetic acid and ten grams of the fine metallic powder, iron and zinc, are placed in a flask of a capacity of about three-quarters of a liter and twenty-five cubic centimeters of the solution of the nitrate added. The flask is covered during the reduction to prevent loss by spraying, and after the solution is complete, which is the case in about ten minutes, the contents of the flask are diluted with from 200 to 300 cubic centimeters of water, thirty cubic centimeters of caustic soda of 1.25 specific gravity added, and the whole distilled as in the kjeldahl process. It must be noted that it is essential that the iron be finely divided; it is mixed with the powdered zinc in equal parts. The total nitrogen can be determined in guanos and nitrate mixtures by the following simple alteration in procedure: One gram of [Pg 231] the substance is dissolved in water, five cubic centimeters of glacial acetic acid, and from two to three grams of the mixed metallic powder added, and the whole gently heated for ten or fifteen minutes. After the contents of the flask have cooled, twenty-five cubic centimeters of sulfuric acid are cautiously added in small portions, undue frothing being restrained by the addition of a fragment of paraffin wax. The acetic acid is then driven off by heating, and the remaining contents of the flask boiled until the organic matter is completely decomposed, as in the kjeldahl process. About two hours boiling is required. Neutralization and distillation are then practiced as in the ordinary manner. The method is also applicable to the determination of nitrates in drinking water, provided nitrites and ammonia be absent.
209. Krüger’s Method for Nitric Acid.—About three-tenths gram of the substance dissolved in water is mixed with twenty cubic centimeters of a hydrochloric acid solution of stannous chlorid holding 150 grams of tin per liter.[177] One and a half grams of spongy tin prepared by the action of zinc on stannous chlorid are added. The flask containing the mixture is heated until the tin is dissolved, by which time the nitric acid is completely reduced. The subsequent distillation and titration are accomplished as usual. In the case of nitro and nitroso compounds, after the solution of the tin, twenty cubic centimeters of sulfuric acid are added and heated until sulfuric vapors escape. After cooling, the amido substances formed are oxidized by potassium bichromate before the distillation takes place.
Krüger also estimates the nitrogen in benzol, pyridin, and chinolin derivatives by dissolving them in sulfuric acid, using from two-tenths to eight-tenths of a gram of the alkaloidal bodies and, after cooling the solution, oxidizing by adding finely powdered potassium bichromate.[178] About half a gram more of the potassium bichromate should be used than is necessary for the oxidation of the substances in solution. The entire oxidation does not consume more than from fifteen to thirty minutes.
210. Functions of Sodium Nitrate.—Practically the only form of oxidized nitrogen which is of importance from an agronomic point of view [Pg 232] is sodium nitrate, often known in commerce by the name Chile saltpeter. Applied to a growing crop it at once becomes dissolved at the first rainfall or by the natural moisture of the soil. It carries thus to the rootlets of plants a supply of nitrogen in the most highly available state. There is perhaps no other kind of plant food which is offered to the living vegetable in a more completely predigested state, and none to which a quicker response will be given. By reason of its high availability, however, it must be used with care. A too free use of such a stimulating food may have, in the end, an injurious effect upon the crop, and is quite certain to lead to the waste of a considerable portion of expensive material. For this reason sodium nitrate should be applied with extreme care, in small quantities at a time and only when it is needed by the growing crop. It would be useless, for instance, to apply this fertilizer in the autumn with the expectation of its benefitting the crop to a maximum degree the following spring. Again, if the application of this salt should be made just previous to a heavy rain almost or quite the whole of it would be removed beyond the reach of the absorbing organs of the plant.
When once the nitric acid has been absorbed by the living rootlet it is held with great tenacity. Living plants macerated in water give up only a trace of nitric acid, but if they be previously killed with chloroform the nitric acid they contain is easily leached out.
The molecule of sodium nitrate is decomposed in the process of the absorption of the nitric acid. The acid enters the plant organism and the soda is excreted and left to combine with the soil acids. The nascent soda may thus play a role of some importance in decomposing particles of minerals containing potash or phosphoric acid. It is probable that the decomposition of the sodium nitrate takes place in the cells of the absorbing plant organs for it is difficult to understand how it could be accomplished externally. While the soda therefore is of no importance as a direct plant food it can hardly be dismissed as of no value whatever in the process of fertilization. Many of the salts of soda as, for instance, common salt, are quite hygroscopic and serve to attract moisture from the air and thus become [Pg 233] carriers of water between the plant and the air in seasons of drought; and sodium nitrate itself is so hygroscopic as not to be suited to the manufacture of gunpowder.
To recapitulate: The chief functions of sodium nitrate are to give to the plant a supply of oxidized nitrogen ready for absorption into its tissues and incidentally to aid, by the residual soda, in the decomposition of silt particles containing potash or phosphoric acid and in supplying to the soil salts of a more or less deliquescent nature.
211. Commercial Forms of Chile Saltpeter.—The Chile saltpeter of commerce may reach the farmer or analyst in the lumpy state in which it is shipped or as finely ground and ready for application to the fields. Unless the farmer is provided with means for grinding, the latter condition is much to be preferred. It permits of a more even distribution of the salt and thus encourages economy in its use. For the chemist also it is advantageous to have the finely ground material, which condition permits more easily a perfect sampling, a process which, with the unground salt, is attended with no little difficulty.
212. Percentage of Nitrogen in Chile Saltpeter.—Chemically pure sodium nitrate contains 16.49 per cent of nitrogen. The salt of commerce is never pure. It contains moisture, potash, magnesia, lime, sulfur, chlorin, iodin, silica and insoluble materials, and traces of other bodies. The value of the salt depends, therefore, not only on the market value of nitrogen at the time of sale, but also on its content of nitrogen. The nitrate of commerce varies greatly in its nitrogen content and is sold on a guaranty of its purity. The best grades range in nitrogen from fifteen to sixteen per cent. The content of nitrogen has long been estimated in the trade by determining the other constituents and counting the rest as nitrogen. This practice arose in former times when no convenient method was at hand for determining nitric nitrogen. The process is tiresome and unreliable because all errors of every kind are accumulated in the nitrogen content, but inasmuch as the method is still required by many merchants, the analyst should be acquainted with it, and it is therefore given further along. The usual methods for determining nitric nitrogen may be applied in all [Pg 234] cases where samples of sodium nitrate are under examination, but some special processes are added for convenience.
213. Adulteration of Chile Saltpeter.—The analyst is the only protector of the farmer in guarding against the practice of adulteration of sodium nitrate aside from the honesty of the dealer. Even the honest dealer is compelled to protect himself against fraud, and therefore, the world over, commerce in this fertilizer is now conducted solely on the analyst’s certificate. Happily, therefore, adulteration is almost unknown because it is certain to be detected. Formerly, the saltpeter was adulterated with common salt, or low grade salts from the potash mines; but it is an extremely rare thing now to find any impurities in the salts other than those naturally present.
In every case the analyst may grow suspicious when he finds the content of nitrogen in a sample to fall below thirteen per cent. It must not be forgotten, however, that some potassium nitrate may be present in the sample, and since that salt contains only 13.87 per cent of nitrogen its presence would tend to lower the value of the fertilizer; but although the potash itself is a fertilizer of value it is not worth more than one-third as much as nitrogen. In all cases of suspected adulteration, it is advisable to make a complete analysis. The results of this work will, as a rule, lead the analyst to a correct judgment.
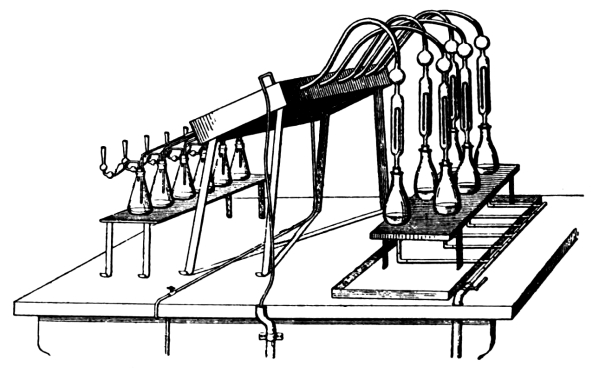
Figure. 15.
Halle Nitric Acid Apparatus.
[Pg 235] 214. The Halle Zinc-Iron Method.—For determining the nitrogen in Chile saltpeter the reduction method is conducted at the Halle Station as follows:[179] Ten grams of the nitrate are dissolved in one liter and fifty cubic centimeters of the solution corresponding to half a gram of the sample, taken for each determination. The apparatus employed is shown in Fig. 15. A mixture of five grams of zinc dust and an equal weight of iron filings is employed as the source of hydrogen. The reduction takes place in an alkaline medium secured by adding to the other materials mentioned, eighty cubic centimeters of soda-lye of 1.30 specific gravity. The respective quantities of iron and zinc may be measured instead of weighed, as exact proportions are not required. After the addition of all the materials the flask is allowed to stand for an hour at room temperature. The distillation is then commenced and continued until at least 100 cubic centimeters of distillate have been collected. The receiving flasks are ordinary erlenmeyers, each of which contains twenty cubic centimeters of set sulfuric acid, as in the usual kjeldahl process. The flasks are sealed with a few drops of water by the device shown in the figure. After the end of the operation the water in each one is washed back into its proper flask with freshly boiled water. During the vigorous evolution of hydrogen, at the beginning of the operation, some kind of a safety arrangement is necessary to prevent the particles of soda-lye being carried over by the bubbles of that gas. The siphon bulb shown in the figure is found effective for this purpose. In this operation better results are obtained by condensing the escaping steam, and for this reason the block tin tubes are conducted through a tank supplied with a current of cold water. The ends of the tubes should not dip below the surface of the liquid in the receivers. When the condensed liquid collects in considerable quantities in the safety tube the lamp should be extinguished under the flask, which permits the return of the liquid to the flask by means of the siphon. This should be done two or three times during the progress of the distillation to prevent a too high concentration of the soda-lye, thus endangering the flask. The excess of the acid in the receiver is determined by titration, as in the regular kjeldahl method. Blank determinations should be made, from time to time, and corrections made in harmony therewith. [Pg 236]
215. Method of the French Sugar Chemists.—The nitrogen in Chile saltpeter is estimated by the French chemists according to the method of Schlösing, described in the first volume, page 500. In order to avoid the trouble of calculating the results from the volume of nitric oxid obtained, a determination is first made with a pure salt, sodium or potassium nitrate. The volume of gas obtained is read directly without correction and taken for direct comparison. The comparison is made as follows:
The solutions of the pure salts and of the sample to be analyzed are made of such a strength as to contain sixty-six grams of sodium nitrate, or eighty grams of potassium nitrate, in a liter. Five cubic centimeters of such a solution will yield a little less than 100 cubic centimeters of nitric oxid under usual conditions. Let the volume of gas obtained with the pure salt be v; and that with the sample be v′. The calculation is then made from the equation:
| v′ | = | x |
| v | 100 |
Example: Let ninety-five cubic centimeters be the volume of gas from five cubic centimeters of the pure salt (sodium nitrate), and 91.5 cubic centimeters be the volume of gas from five cubic centimeters of the sample; then
| 91.5 | = | x | , whence x = 96.31. |
| 95 | 100 |
Hence the sample analyzed contains 96.31 per cent of sodium nitrate. Since the pure sodium nitrate contains 16.47 per cent of nitrogen the sample under examination would contain
| 16.47 × 96.31 | = 15.86 per cent. |
| 100 |
It is evident that this comparative method is quite easy of application when the sample under examination has no other nitrate in it except that combined with the one base.
216. Volumetric Method of Gantter.—The process proposed by Gantter for determining the nitrogen, volumetrically, in Chile saltpeter and other nitrates is based on the following principles:[180]
(1) If a nitrate be heated in contact with sulfuric and phosphorous acids, nitrous acid will be formed.
(2) If nitrous acid be boiled with ammonium chlorid, nitrogen will be quantitatively evolved from both compounds. These processes are illustrated by the following formulas: [Pg 237]
(a) N₂O₅ + P₂O₃ = N₂O₃ + P₂O₅.
(b) N₂O₃ + 2NH₄Cl = 2N₂ + 3H₂O + 2HCl.
Figure. 16.
Gantter’s Nitrogen Apparatus.
It is seen from the above that the nitrate will give, by this treatment, double the volume of nitrogen which it contains. In practice, the two reactions may be secured in one operation by warming the nitrate solution slowly with sulfuric and phosphorous acids and ammonium chlorid. The nitric acid, as it becomes free, gives a part of its oxygen to the phosphorous compound, and the nitrous acid, in a nascent state, is at once reduced by the ammonium chlorid. There are two sources of error which must be guarded against in the work; a portion of the nitrogen may escape reduction to the elementary state, or some of the nitrate may fail to be decomposed. These errors are easily avoided if the reaction be begun slowly, so that the evolution of gas may be gradual. The temperatures at first should, therefore, be kept as low as possible. The development of red fumes, showing the presence of undecomposed nitrogen oxids, shows that the results will be too low. It is necessary, also, to provide for the absorption of the hydrochloric acid which is formed. The reaction is very conveniently conducted in the apparatus shown in Fig. 16. The decomposition takes place in the flask A and the mixed gases pass into the absorption bulb C. The delivery-tube is very much expanded, as shown in the figure, so [Pg 238] that no soda-lye can enter A during the cooling of the flask. The absorption bulb is connected with A and B by the tubes a and b as shown. The tube d connects the apparatus with the gasvolumeter.[181] The bulb B serves as a pipette for the introduction of the decomposing acid. The operation is conducted as follows: Three cubic centimeters of the nitrate solution, containing no more than 300 milligrams of substance, are placed in the flask A with half a gram each of crystallized ammonium chlorid and phosphorous acid. In the bulb B are placed seven cubic centimeters of sulfuric acid to which has been added one-third its volume of water. Two cubic centimeters of acid are allowed to flow from B into A. The apparatus is brought to a constant temperature by being immersed in a large cylinder, E, containing water at a temperature which can easily be controlled. When this constant temperature has been reached the apparatus is taken from the cooling cylinder which contains also a smaller cylinder, D, nearly filled with water and connected through f′ with the measuring apparatus M. The barometer-tube F is half filled with colored water so that the pressure may be equalized before and after the operation. The flask A is warmed very gently at first, and the nitrogen evolved is conducted into D driving an equivalent volume of water into M. The evolution of the gas must be carefully controlled and the heat at once removed if it become too rapid. The appearance of a red color shows the evolution of oxids of nitrogen rendering the analysis inexact. When the evolution of nitrogen has nearly ceased the lamp is removed and some more sulfuric acid allowed to flow into A from B, after which A is again heated, this time to the boiling-point. All vapors of hydrochloric acid produced are absorbed by the soda-lye in C. The boiling is continued a few minutes, but not long enough to darken the liquid in A. After replacing the apparatus in the cylinder E and bringing both temperature and pressure to the same point as before the beginning of the operation, the volume of nitrogen evolved is determined by measuring the water in M.
The apparatus is first set by using pure potassium or sodium nitrate. Since the temperature and pressure do not vary much within an hour or two the volume of water obtained with a sample of white saltpeter can be [Pg 239] compared directly with that given off by the same weight of a pure potassium or sodium nitrate without correction.
Example.—Two hundred and fifty milligrams of potassium nitrate, containing 34.625 milligrams of nitrogen, displaced in a given case sixty cubic centimeters of water; therefore one cubic centimeter of water equals 0.578 milligram of nitrogen. If 289 instead of 250 milligrams be taken then the number of cubic centimeters of water displaced divided by five will give the per cent of nitrogen.
217. Method of Difference.—In the analysis of Chile saltpeter by the direct method a variation of 0.25 per cent in the content of nitrogen is allowed from the dealers’ guaranty. This would allow a total variation in the content of sodium nitrate of 1.52 per cent. Dealers and shippers have always been accustomed to estimate the quantity of sodium nitrate in a sample by difference; i. e., by estimating the constituents not sodium nitrate and subtracting the sum of the results from 100. Chile saltpeter usually contains sodium nitrate, water, insoluble ferruginous matters, sodium chlorid, sodium sulfate, magnesium chlorid, sodium iodate, calcium sulfate and sometimes small quantities of potassium nitrate.
When the total sodium nitrate is to be estimated by difference the following procedure, arranged by Crispo,[182] may be followed:
Water.—Dry ten grams of the finely powdered sample to constant weight at 150°-160°.
Chlorin.—The residue, after drying, is dissolved and the volume made up to one-fourth liter with water and the chlorin determined in one-fifth thereof and calculated as sodium chlorid.
Insoluble.—Twenty grams are treated with water until all soluble matter has disappeared, filtered on a tared gooch, and the filtrate dried to constant weight.
Sulfuric Acid.—The sulfuric acid is precipitated by barium chlorid in the slightly acid filtrate from the insoluble matter. The acidity is produced by a few drops of nitric acid. The rest of the process is conducted in the usual way.
Magnesia.—This is precipitated by ammonium sodium phosphate, filtered, ignited, and weighed as pyrophosphate. The magnesium is then [Pg 240] calculated as chlorid. Magnesia is rarely found in excess of one-fourth per cent. When this amount is not exceeded the estimation of it may be neglected without any great error. As has already been said the chlorin is all calculated as sodium chlorid. If a part of it be combined with one-fourth per cent of magnesia it would represent 0.59 per cent of magnesium chlorid instead of 0.73 per cent sodium chlorid. In omitting the estimation of the magnesia therefore the importer is only damaged to the extent of 0.14 per cent of sodium nitrate.
Sodium Iodate.—This body, present only in small quantities, may also be neglected. In case the content of this body should reach one-fourth per cent the estimation of chlorin by titration using potassium chromate as indicator is impracticable. Such an instance, however, is rarely known.
Approximate Results.—When the determinations outlined above have been carefully made it is claimed that the result obtained by subtraction from 100 will not vary more than from two-tenths to three-tenths per cent from the true content of sodium nitrate. The method, however, cannot be considered strictly scientific and is much more tedious and chronophagous than the direct determination. In the direct determination, however, the analyst must assure himself that potassium is present in only appreciable quantities otherwise the per cent of sodium nitrate will be too low.
The presence of potassium nitrate is a detriment in this respect only; viz., that it contains a less percentage of nitrogen than the corresponding sodium salt. As a fertilizer, the value of Chile saltpeter may be increased by its content of potassium.
218. The Application of Chile Saltpeter to the Soil.—The analyst is often asked to determine the desirability of the use of sodium nitrate as a fertilizer and the methods and times of applying it. These are questions which are scarcely germane to the purpose of this work but which, nevertheless, for the sake of convenience may be briefly discussed. In the first place it may be said that the data of a chance chemical analysis will not afford a sufficiently broad basis for an answer. A given soil may be very rich in nitrogen as revealed by chemical analysis, and yet poor in an available supply. This is [Pg 241] frequently the case with vegetable soils, containing, as they do, large quantities of nitrogen but holding it in practically an inert state. I have found such soils very rich in nitrogen, yet almost entirely devoid of nitrifying organisms. It is necessary therefore in reaching a judgment on this subject from analytical data to consider the different states in which the nitrogen may exist in a soil and above all the nitrifying power of the soil if the nitrogen be chiefly present in an organic state. Culture solutions should therefore be seeded with samples of the soil under examination and the beginning and rapidity of the nitrification carefully noted. In conjunction with this the nitrogen present in the soil in a nitric or ammoniacal form should be accurately determined. These determinations should be made according to the directions given in the first volume, pp. 448-548.
For the determination of nitrifying power we prefer the following method:
219. Taking Samples of Soil in Sterilized Tubes.—Brass tubes are prepared twenty centimeters in length and one and a half in diameter. One end is ground to a beveled edge and compressed in a mold so as to make the cutting edge slightly smaller in diameter than the internal diameter of the tube. It is then ground or filed until smooth and sharp. The blunt end of the tube is stoppered loosely with cotton and it is then sterilized by heating for an hour to 150°. Rubber caps are provided and each one has placed at the bottom a rubber ball to prevent the rubber from being cut by the edges of the brass tube. The caps should be of two distinct colors. Half of the rubber caps are sterilized by being boiled for an hour in water for three successive days. The caps cannot be heated to 150° dry heat with safety. On removing the brass tube from the sterilizing oven as soon as it is cool enough to handle, a sterilized rubber cap is slipped over its cutting end. An unsterilized cap is then slipped over the other end containing the cotton plug. Inasmuch as the cotton plug is never removed it is not necessary to sterilize the cap covering it. Large numbers of the tubes can thus be prepared for use and they can be safely transmitted to a distance by express or mail. For convenience, each tube is encased in a small cloth bag, which is tied with a cord carrying a tag on which the [Pg 242] necessary data can be recorded at the time of taking the sample.
The tubes and their rubber caps thus carefully sterilized should not be removed from their cloth envelopes until the moment of taking each sample. After the sample has been taken and the cap replaced on the tube the latter should be immediately enclosed in the cloth sack and labeled with one of the tags therewith enclosed. The sample should be taken in two kinds of soil, in one instance in a cultivated soil, which is most characteristic of the locality, and in the second place a virgin soil of the same type. The virgin soil may be either soil which has been covered with grass or in forest. The spots at which the samples are to be taken having been previously selected, the tags for each tube should be prepared beforehand so as to avoid delay at the time of sampling. A pit with straight walls should be dug, the sides of which are at least two feet wide and even three feet would be better. The pit should be about forty-two inches deep. One of the sides having been made perfectly smooth and without allowing the loose fragments from the top to fall down and adhere to the walls below, the spots at which the samples are to be taken should be marked with a tape line at the following points; viz., three, fifteen, twenty-seven, and thirty-nine inches, respectively, below the surface. Beginning at the bottom point, carefully scrape off the surface of the wall over an area slightly larger than that of the end of the sample tube by means of a spatula, which, just previous to use, has been held for a moment or two in the flame of an alcohol or other convenient lamp. The sample tube having been removed from its sack, it will be noted that the end covered with black rubber is the one which is to be held in the hand, and this black rubber cap should be first removed being careful not to extract the plug of sterilized cotton which closes the end of the tube. Holding the tube firmly by the end, the fingers extending only about two inches from the end, remove the light-colored cap and push the tube with a turning motion into the side of the pit at the point where the surface has been removed with the sterilized spatula. When this is properly done the tube will be filled with a cylinder of soil equal to the length of the part of the tube penetrating the wall of the pit. [Pg 243] The tube is then withdrawn, the light rubber cap first replaced, and then the black one. The light rubber cap should be held in the hand during the process in such a way that no dust or particles of soil are permitted to contaminate its inner walls. For this purpose the open end of the cap should be held downward. For the same reason after the removal of the light rubber cap the brass tube should be carefully preserved from dust or fragments, the open end, that is the cutting end, being held downward until ready for use. After one tube has been filled, capped, replaced in the sack and labeled, the spatula should be again sterilized and samples taken in regular order until the top one is finished.
220. Directions for Taking Bulk Samples.—From the sides of the pit described above, bulk samples should be taken as follows:
By means of a spade the soil should be removed from the four sides to the depth of six or nine inches or until the change of color between the soil and subsoil is noted; in all enough to make about 150 pounds of the air-dried soil. In the same way take a sample of the subsoil to the depth of nine additional inches. Remove all stones, large pebbles, sticks, roots, etc., and spread the samples in a sheltered place where they can be air-dried as rapidly as possible. The bulk samples should be taken both from the cultivated and virgin soils. In selecting the cultivated soil, preference should be given to those soils which have not been fertilized within a few years. If recent fertilization have been practiced the character and amount of it should be noted.
221. The Nitrifiable Solution.—The solution to test the nitrifying power of the samples collected as above described is conveniently made as follows:
One liter of the above solution is enough for ten samples, each of 100 cubic centimeters. This quantity is placed in an erlenmeyer, which is stoppered with cotton and sterilized by being kept at 100° for an [Pg 244] hour on three successive days. The erlenmeyer should be sterilized beforehand by heating for an hour at 150°. The freshly precipitated and washed calcium carbonate should be sterilized separately and added to each erlenmeyer at the time of seeding. Enough should always be used to be in excess of the nitrous and nitric acids found. The seeding is accomplished by filling a sterilized spoon which holds approximately half a gram of the soil, from the contents of one of the brass tubes, lifting the plug in the erlenmeyer and transferring quickly to the flask. This should be done in a perfectly still room, preferably as high above the ground as possible and in a place free from dust and under cover. The cotton plug being replaced the erlenmeyer is shaken until the sample of soil added is thoroughly disintegrated and intimately mixed with its contents. With care and experience the seeding is easily accomplished without danger of accidental contamination.
At the end of each period of five days the beginning and progress of nitrification should be determined by some of the methods described in volume first. Either the ammonia can be determined by nesslerizing or the nitrous and nitric acids estimated. For nitrous acid we prefer the method described in volume first, paragraph 504, and for nitric the one in same volume paragraphs 497 and 498.
By supplementing the analysis of a soil by the above described experiments in nitrification the analyst will be able to judge with sufficient accuracy of its needs for nitric nitrogen.
222. Quantity of Chile Saltpeter to be Applied.—The quantities of Chile saltpeter which should be applied per acre vary with so many conditions as to make any definite statement impossible. On account of the great solubility of this salt no more should be used than is necessary for the nutrition of the crop. For each 100 pounds used, from fourteen to fifteen pounds of nitrogen will be added to the soil. Field crops, as a rule, will require less of the salt than garden crops. There is an economic limit to the application which should not be passed. As a rule 250 pounds per acre will prove to be a maximum dressing. The character of the crop must also be considered. Different amounts are required for sugar beets, tobacco, wheat, and other standard [Pg 245] crops. It is rarely the case that a crop demands a dressing of Chile saltpeter alone. It will give the best effects, as a rule, when applied with phosphoric acid or potash. But this is a branch of the subject which cannot be entered into at greater length in this manual. The reader is referred to Stutzer’s work on Chile saltpeter for further information.[183]
223. Consumption of Chile Saltpeter.—The entire consumption of sodium nitrate for manurial purposes in the whole world for 1894 was 992,150 metric tons, valued at $41,000,000. For the several countries using it the consumption was distributed as follows:
| Germany | 397,200 | tons. |
| France | 187,100 | “ |
| England | 117,000 | “ |
| Belgium | 123,000 | “ |
| United States | 100,000 | “ |
| Holland | 56,700 | “ |
| Italy and Spain | 5,200 | “ |
| Other countries | 5,950 | “ |
| Total | 992,150 | “ |
The above figures represent the actual commerce of each country in Chile saltpeter, and may not give the exact consumption.[184] For instance, Germany exports sodium nitrate to Russia and Austria, but it imports this salt from Holland and Belgium. Belgium imports from France, but its exportation is greater than its importations from that country, so that its actual consumption on the farm probably falls considerably below that given on the table. Holland also exports larger quantities than are imported from neighboring states. The exports from England are inconsiderable compared with the quantities received, amounting only to about 5,000 tons a year, while the exportations from France reach nearly 10,000 tons.
Sodium nitrate has a moderate value at the factories where it is prepared for shipment in Chile. Its chief value at the ports where it is delivered for consumption comes from freights and profits of the syndicate. The factories, where it is prepared for the market, are at or near the deposits, and the freights thence to the sea coast are very high. The rail roads which have been constructed to the high plateaux [Pg 246] which contain the deposits, have been built at a very great cost, and the freights charged are correspondingly high. There is also a tax of $1.20 levied on each ton exported. Deducting all costs of transportation and export duties the actual value of sodium nitrate at the factory, ready for shipment, is about sixteen dollars in gold a ton.
[125] American Chemical Journal, Vol. 13, No. 7.
[126] Atwater: Report of the U. S. Commissioner of Fish and Fisheries, 1888, pp. 679-868.
[127] American Naturalist, Vol. 14, p. 473.
[128] Wiley: Retiring Address as President of American Chemical Society, Baltimore Meeting, Dec. 1893, Journal of the American Chemical Society, Vol. 16, pp. 17-20.
[129] Vid. op. et. loc. cit. supra.
[130] Vid. op. et. loc. cit. 4.
[131] Vid. op. et. loc. cit. 4.
[132] Vid. op. et. loc. cit. 4.
[133] Bulletin No. 43, p. 343 (to paragraph 156).
[134] American Chemical Journal, Vol. 2, pp. 27, et seq.
[135] Vid. op. et. loc. cit. supra.
[136] Weather Bureau: Barometers and Measurements of Atmospheric Pressures.
[137] Physikalisch-Chemische Tabellen Landolt und Börnstein, S. 32.
[138] Battle and Dancy: Conversion Tables, p. 34.
[139] Bulletin No. 43, p. 348 (to paragraph 169).
[140] Guide pour le dosage de l’Azote No. 3, p. 8.
[141] Zeitschrift für analytische Chemie, Band 23, S. 557.
[142] Chemiker Zeitung, Band 8, S. 1747.
[143] Journal of the Chemical Society, Transactions, 1881, p. 87.
[144] Bulletin No. 16, p. 51.
[145] Bulletin No. 28, p. 193 (to paragraph 175).
[146] Journal Society of Chemical Industry, Vol. 2, p. 21.
[147] Bulletin de l’Association des Chimistes de Sucrèrie, Tome 9, p. 598.
(bis p. 192) Journal Chemical Society; Transactions, Vol. 21, p. 161.
[148] Zeitschrift für analytische Chemie, Band 22, S. 366.
[149] Vid. op. cit. supra, pp. 370, et seq.
[150] Vid. op. cit., 24. Band 24, S. 455.
[151] Chemisches Centralblatt, 1886, S. 165.
[152] Nederlandsche Staatscourant, Jan. 11, 1893.
[153] Die Agricultur-Chemische Versuchs-Station, Halle a/S., S. 34.
(bis. p. 204) Bulletin No. 43, p. 345.
[154] Bulletin No. 31, 142.
[Pg 247]
[155] Fresenius, quantitative Analyse, 6th Auflage, S. 731.
[156] Zeitschrift für Analytische Chemie, Band 24, S. 455.
[157] Vid. op. cit. supra, Band 25, S. 149.
[158] Archive der Pharmacie {3}, Band 23, S. 177.
[159] Chemisches Centralblatt, 1886, S. 375.
[160] Vid. op. cit. supra, S. 161.
[161] Vid. op. cit. 35, S. 433.
[162] Vid. op. et. loc. cit. 28.
[163] Vid. op. cit. 29, S. 44.
[164] Bulletin No. 16, p. 51.
[165] Zeitschrift für analytische Chemie, Band 28, S. 188.
[166] Bulletin No. 35, p. 68.
[167] Bulletin No. 35, p. 202.
[168] Bulletin No. 112, Connecticut Agricultural Experiment Station, and Bulletin No. 35, p. 96.
[169] Guide pour le dosage de l’Azote, p. 14.
[170] Revue de Chimie Analytique Appliquée, Tome 1, p. 51.
[171] Chemiker Zeitung, Band 4, S. 360.
[172] Bulletin No. 43, p. 361.
[173] Bulletin de la Société Chimique, 1890, p. 324.
[174] Bulletin No. 35, p. 88.
[175] Die Agricultur-Chemische Versuchs-Station, Halle a/S., S. 50.
[176] Vid., Vol. 1, p. 539.
[177] Berichte der deutschen chemischen Gesellschaft, Band 27,S. 1633.
[178] Vid. op. et. loc. cit. supra.
[179] Vid. op. cit. 51, S. 48.
[180] Zeitschrift für analytische Chemie, Band 34, S. 26.
[181] Vid. op. cit. supra, Band 32, S. 553.
[182] L’Engrais, Tome 9, p. 877.
[183] Der Chile Saltpeter; Seine Bedeutung und Anwendung als Düngemittel.
[184] L’Engrais, 5 Avril, 1895, p. 324.
[Pg 248]
224. Introduction.—The potash present in unfertilized soils has been derived from the decay of rocks containing potash minerals. Among these potash producers feldspars are perhaps the most important. For a discussion of the nature of their decomposition and the causes producing it the first part of volume first may be consulted. Potash is quite as extensively distributed as phosphoric acid and no true soils are without it in some proportion. Its presence is necessary to plant growth and it forms, in combination with organic and mineral acids, an essential part of the vegetable organism, existing in exceptionally rich quantities in the seeds. It is possible that potash salts, such as the chlorid, sulfate, and phosphate may be assimilated as such, but, as with other compounds, we must not deny to the plant the remarkable faculty of being able to decompose its most stable salts and to form from the fragments thus produced entirely new compounds. This is certainly true of the potash compounds existing in plants in combination with organic acids. The potash which is assimilated by plants exists in the soil chiefly in a mineral state, and that added as fertilizer is chiefly in the same condition. That part of the potash in a soil arising directly from the decomposition of vegetable matters may exist partly in organic combination, but this portion, in comparison with the total quantity absorbed by the plant, is insignificant.
It is then safe to assume that at least a considerable part of the potash absorbed by the plant is decomposed from its original form of combination by the vegetable biochemical forces, and is finally incorporated in the plant tissues in forms determined by the same powerful forces of vegetable metabolism.
The analyst is not often called upon to investigate the forms in which the potash exists in plants, when engaged in investigation of fertilizers. It is chiefly found in combination with organic and [Pg 249] phosphoric acids, and on ignition will appear as phosphate or carbonate in the ash.
225. Forms in which Potash is Found in Fertilizers.—The chief natural sources of potash used in fertilizer fabrication are: First, organic compounds, such as desiccated mineral matters, tobacco waste, cottonseed hulls, etc.; second, the ash derived from burning terrestrial plants of all kinds; third, the natural mineral deposits, such as Stassfurt salts.
All of these forms of potash may be found in mixed fertilizers. While the final methods of analyses are the same in all cases the preliminary treatment is very different, being adapted to the nature of the sample. For analytical purposes, it is highly important that the potash be brought into a soluble mineral form, and that any organic matters which the sample contains be destroyed. If the sample be already of a mineral nature, it may still be mixed with other organic matter and then it requires treatment as above, for it is not safe always to rely solely on the solubility of the potash mineral, and the solution, moreover, in such cases, is likely to contain organic matter. In some States, only that portion of the potash soluble in water is allowed to be considered in official fertilizer work. In these cases it is evident that the organic matter present should not be destroyed in the original sample, but only in the aqueous solution. Since, however, the potash occluded in organic matter becomes constantly available as the process of decay goes on, it is not just to exclude it from the available supply. It may not be so immediately available as when in a soluble mineral state, but it is not long before it becomes valuable. Experience has shown, moreover, that phosphorus, nitrogen, and potash are all more valuable finally when applied to the soil in an organic form. This fact is a corroboration of the theory already advanced that all mineral compound bodies are probably decomposed before they enter as component parts into the tissues of the vegetable organism.
It is highly probable, therefore, that the potash existing in organic compounds, finely divided and easily decomposed, is of equal, if not greater value to plant life than that already in a soluble mineral state. The organic matter, when present, is destroyed, either by ignition at a low temperature, or by moist combustion with an oxidizing agent before the potash is precipitated. [Pg 250]
226. Tobacco Stems and Waste.—Until within a few years tobacco stems and other waste from factories, were treated as a nuisance in this country, and burned or dumped into streams. By burning and saving the ash the potash contained in the stems and waste would be recovered in a form suitable for field use. The nitrogen, however, contained in these waste materials, both in the form of nicotin and of albuminoids would be lost. Ignition of this waste, therefore, should not be practiced. It should be prepared for use by grinding to a fine powder. Applied to the soil in this condition the powder may be useful as an insecticide as well as a fertilizer. Tobacco stems contain from twelve to twenty-seven per cent of moisture, and from twelve to twenty per cent of ash. The composition of the stems from two celebrated tobacco growing regions is subjoined:[185]
| Kentucky stems. | Connecticut stems. | ||||||
|---|---|---|---|---|---|---|---|
| Moisture | 26.70 | per | cent. | 13.47 | per | cent. | |
| Organic and volatile | 60.18 | “ | “ | 70.85 | “ | “ | |
| Ash | 13.12 | “ | “ | 15.68 | “ | “ | |
The ash calculated to the original substance had the following composition:
| Kentucky stems. | Connecticut stems. | ||||||
|---|---|---|---|---|---|---|---|
| Phosphoric acid | 0.67 | per | cent. | 0.53 | per | cent. | |
| Potash | 8.03 | “ | “ | 6.41 | “ | “ | |
It is thus seen that about half the ash of tobacco stems is composed of potash. The stalks of the tobacco have almost the same composition as the stems, but the percentage of ash is not quite so great. In three samples analyzed at the Connecticut station the percentages of ash found in the water-free substance were 6.64, 7.00, and 7.46 respectively. The pure ash of the stalks was found to have the following composition:[186]
| Description of samples. | |||||||
|---|---|---|---|---|---|---|---|
| Constituents | Cut Aug. 22. | Cut Sept. 17. | |||||
| Silica | 0.82 | per | cent. | 0.57 | per | cent. | |
| Iron and aluminum oxids | 1.38 | “ | “ | 1.38 | “ | “ | |
| Lime | 14.01 | “ | “ | 16.58 | “ | “ | |
| Magnesia | 6.64 | “ | “ | 7.36 | “ | “ | |
| Potash | 56.34 | “ | “ | 54.46 | “ | “ | |
| Soda | 1.28 | “ | “ | 1.16 | “ | “ | |
| Sulfuric acid | 8.06 | “ | “ | 6.75 | “ | “ | |
| Phosphoric acid | 6.37 | “ | “ | 6.27 | “ | “ | |
| Chlorin | 6.55 | “ | “ | 7.05 | “ | “ | |
| 101.45 | “ | “ | 101.58 | “ | “ | ||
| Deduct oxygen = chlorin | 1.45 | “ | “ | 1.58 | “ | “ | |
| 100.00 | “ | “ | 100.00 | “ | “ | ||
[Pg 251] The leaves of the tobacco contain more ash than the stalks or stems, but the percentage of potash therein is less. In eighteen samples analyzed at the Colorado station the percentages of moisture in the leaf varied from 6.08 to 28.00, and those of ash from 22.60 to 28.00.[187] The percentages of potash in the ash varied from 15.20 to 26.30. In these data the carbon dioxid, sand, etc., are included, while in those quoted from the Connecticut station they were excluded.
227. Cottonseed Hulls and Meal.—A considerable quantity of potash is added to the soil in cottonseed meal and hulls. The practice of burning the hulls cannot be recommended, although it is frequently practiced, for the incineration does not increase the quantities of phosphoric acid and potash, while it destroys the availability of the nitrogen. Nevertheless the analyst will often have to deal with samples of the raw materials above mentioned, as well as with the ash of the hulls, in which the potash can be determined by some one of the standard methods to be described. In general it is found that the hulls of seeds and the bark and leaves of plants have a greater percentage of ash than the interior portions. In the case of cottonseed however, an exception is to be noted. The cottonseed meal in the air-dried state has about seven per cent of ash, while the hulls have only about three. When it is remembered, however, that the greater part of the oil has been removed from the meal it will be seen that in the whole seed in the fresh state the discrepancy is not so marked.
In the crude ash of the hulls the percentage of potash varies generally from twenty to twenty-five per cent, but in numerous cases these limits are exceeded. In twelve samples of cottonseed hull ashes examined by the Connecticut station the mean percentage of potash in the crude sample was 22.47, and the extremes 15.57 and 30.24 per cent respectively.[188] In determining the value of the ash per ton the content of phosphoric acid must also be taken into account.
Cottonseed meal contains about 1.75 per cent of potash. Since the mean percentage of ash in the meal is seven, the mean content of potash in the crude ash is about twenty-five.
228. Wood Ashes.—Unleached wood ashes furnish an important quantity of potash fertilizer. The composition of the ash of woods [Pg 252] is extremely variable. Not only do different varieties of trees have varying quantities of ash, but in the same variety the bark and twigs will give an ash quite different in quantity and composition from that furnished by the wood itself. In general the hard woods, such as hickory, oak, and maple, furnish a quality of ash superior for fertilizing purposes to that afforded by the soft woods, such as the pine and tulip trees.
The character of the unleached wood ashes found in the trade is indicated by the subjoined analyses. The first table contains the mean, maximum and minimum results of the analyses of ninety-seven samples by Goessmann.[189]
| Mean composition of wood ashes. | |||
|---|---|---|---|
| Means. | Maxima. | Minima. | |
| Potash | 5.5 | 10.2 | 2.5 |
| Phosphoric acid | 1.9 | 4.0 | 0.3 |
| Lime | 34.3 | 50.9 | 18.0 |
| Magnesia | 3.5 | 7.5 | 2.3 |
| Insoluble | 12.9 | 27.9 | 2.1 |
| Moisture | 12.0 | 28.6 | 0.7 |
| Carbon dioxid and undetermined | 29.9 | ||
In sixteen analyses made at the Connecticut station the data obtained are given below:[190]
| Means. | Maxima. | Minima. | |
|---|---|---|---|
| Potash | 5.3 | 7.7 | 4.0 |
| Phosphoric acid | 1.4 | 1.8 | 0.9 |
In fifteen analyses of ashes from domestic wood-fires in New England stoves, the following mean percentages of potash and phosphoric acid were found:
| Potash | 9.63 |
| Phosphoric acid | 2.32 |
In leaching, ashes lose chiefly the potassium carbonate and phosphate which they contain. Leached and unleached Canada ashes have the following composition:
| Unleached. | Leached. | ||||||
|---|---|---|---|---|---|---|---|
| Insoluble | 13.0 | per | cent. | 13.0 | per | cent. | |
| Moisture | 12.0 | “ | “ | 30.0 | “ | “ | |
| Calcium carbonate and hydroxid | 61.0 | “ | “ | 51.0 | “ | “ | |
| Potassium carbonate | 5.5 | “ | “ | 1.1 | “ | “ | |
| Phosphoric acid | 1.9 | “ | “ | 1.4 | “ | “ | |
| Undetermined | 6.6 | “ | “ | 3.5 | “ | “ | |
In the wood ashes of commerce therefore, it is evident that the proportion of the potash to the lime is relatively low.
The number of parts by weight of the chief ingredients of the ash in ten thousand pounds of woods of different kinds is given in the table below together with the percentage composition of the pure ash, that is the crude ash deprived of carbon and carbon dioxid. [Pg 253]
Pounds of the Ingredients Named
in Ten Thousand Pounds of Wood.
| Dogwood. | Sycamore. | Post oak. | Ash. | Red Oak. | Hickory. | |
|---|---|---|---|---|---|---|
| Cornus florida. |
Platanus occidentalis. |
Q. obtusiloba. | F. Americana. | Q. rubra. | Carya tomentosa. |
|
| Potash | 9.02 | 18.06 | 16.85 | 14.94 | 13.95 | 13.80 |
| Phosphoric acid | 5.72 | 9.55 | 6.96 | 1.15 | 5.98 | 5.83 |
| Lime | 6.41 | 24.73 | 35.61 | 7.60 | 27.40 | 18.40 |
| Magnesia | 14.67 | .49 | 5.28 | .10 | 3.05 | 4.86 |
| White oak. | Magnolia. | Georgia pine. | Yellow pine. | Black pine. | Chestnut. | Old field pine. |
|
|---|---|---|---|---|---|---|---|
| Q. alba. | M. grandiflora. | P. palustris. | P. mitis. | Picea nigra. | Castanea vesca or sativa. |
P. mitis. | |
| Potash | 10.60 | 7.13 | 5.01 | 4.54 | 3.02 | 2.90 | .79 |
| Phosphoric acid | 2.49 | 3.19 | 1.24 | .96 | .92 | 1.09 | .73 |
| Lime | 7.85 | 14.21 | 18.04 | 15.16 | 12.46 | 7.93 | 12.12 |
| Magnesia | .90 | 2.94 | 2.03 | .74 | .10 | .34 | 1.17 |
The Pure Ashes of the Woods
Contain the Following Per Cents
of the Ingredient Named.
| Dogwood. | Sycamore. | Post oak. | Ash. | Red Oak. | Hickory. | |
|---|---|---|---|---|---|---|
| Cornus florida. |
Platanus occidentalis. |
Q. obtusiloba. | F. Americana. | Q. rubra. | Carya tomentosa. |
|
| Potash | 28.04 | 23.17 | 21.92 | 46.04 | 24.66 | 28.60 |
| Phosphoric acid | 8.51 | 12.23 | 9.00 | 3.58 | 10.55 | 11.97 |
| Lime | 38.93 | 31.62 | 46.39 | 23.57 | 48.26 | 37.94 |
| Magnesia | 6.80 | .62 | 6.88 | .60 | 5.38 | 10.04 |
| White oak. | Magnolia. | Georgia pine. | Yellow pine. | Black pine. | Chestnut. | Old field pine. |
|
|---|---|---|---|---|---|---|---|
| Q. alba. | M. grandiflora. | P. palustris. | P. mitis. | Picea nigra. | Castanea vesca or sativa. |
P. mitis. | |
| Potash | 42.16 | 19.54 | 15.35 | 19.70 | 14.30 | 18.10 | 3.85 |
| Phosphoric acid | 9.48 | 8.75 | 3.82 | 4.18 | 4.33 | 6.76 | 4.11 |
| Lime | 29.85 | 38.94 | 55.24 | 65.53 | 58.98 | 49.18 | 67.73 |
| Magnesia | 3.43 | 8.05 | 6.25 | 3.20 | .50 | 2.11 | 6.54 |
[Pg 254] 229. Fertilizing Value of Ashes.—Primarily, the fertilizing value of wood-ashes depends on the quantity of plant food which they contain. With the exception of potash and phosphoric acid, however, the constituents of wood-ashes have little, if any, commercial value. The beneficial effects following the application of ashes, however, are greater than would be produced by the same quantities of matter added in a purely manurial state. The organic origin of these materials in the ash has caused them to be presented to the plant in a form peculiarly suited for absorption. Land treated generally with wood-ashes becomes more amenable to culture, is readily kept in good tilth, and thus retains moisture in dry seasons and permits of easy drainage in wet. These effects are probably due to the lime content of the ash, a property moreover favorable to nitrification and adapted to correcting acidity. Injurious iron salts, which are sometimes found in wet and sour lands, are precipitated by the ash and rendered innocuous or even beneficial. A good wood-ash fertilizer therefore is worth more than would be indicated by its commercial value calculated in the usual way.
230. Molasses from Sugar-Beets.—The residual molasses resulting after the extraction of all the crystallizable sugar in beet-sugar manufacture is very rich in potash. The molasses contains from ten to fifteen per cent of ash.
The composition of the ash varies greatly in the content of potash as well as of the other constituents.[191] The content of potassium carbonate varies from twenty-two to fifty-five per cent and, in addition to this, some potassium sulfate and chlorid are usually present.
The following figures give the composition of a good quality of beet-molasses ash:
| Potassium | carbonate | 45.30 | per cent | |
| Sodium | “ | 13.86 | “ | |
| Potassium | chlorid | 22.40 | “ | |
| “ | sulfate | 8.00 | “ | |
| Silica, lime, alumina, water, phosphoric | ||||
| acid, and undetermined | 15.82 | “ | ||
Thus, in 100 parts of such an ash over three-quarters are potash salts. The molasses may be applied directly to the soil or diluted and sprayed over the fields. [Pg 255]
231. Residue of Wineries.—The pomace of grapes after being pressed or fermented for wine production contains considerable quantities of potash as crude argol or acid potassium tartrate. This material can be applied directly to the soil or first burned, when its potash will be secured in the form of carbonate.
The use of the winery refuse for fertilizing purposes has not assumed any commercial importance in this country.
232. Destruction of Organic Matter by Direct Ignition.—The simplest and most direct method for destroying organic matter is by direct ignition. The incineration may be conducted in the open air or in a muffle and the temperature should be as low as possible. In no case should a low red heat be exceeded. By reason of the moderate draft produced in a muffle and the more even heat which can be maintained this method of burning is to be preferred. With the exercise of due care, however, excellent results can be obtained in an open dish or one partly closed with a lid. At first, with many samples, the organic matter will burn of its own accord after it is once ignited, and during this combustion the lamp should be withdrawn. The ignition in most cases should be continued in a platinum dish but should the sample contain any reducible metal capable of injuring the platinum a porcelain vessel should be used. The lamp should give a diffused flame to avoid overheating of any portions of the dish and to secure more uniform combustion. In using a muffle the heat employed should be only great enough to secure combustion and the draft should be so regulated as to avoid loss due to the mechanical deportation of the ash particles.
233. Ignition with Sulfuric Acid.—The favorable action of sulfuric acid in securing a perfect incineration may also be utilized in the preparation of samples containing organic matter for potash determinations. In this case the bases which by direct ignition would be secured as carbonates are obtained as sulfates. In the method adopted by the official chemists it is directed to saturate the sample with sulfuric acid and to ignite in a muffle until all organic matter is destroyed.[192] Afterwards, when cool the ash is moistened with a little hydrochloric acid and warmed, whereby it is the more easily detached from the dish. [Pg 256] The potash is then determined by any one of the standard methods. This method has several advantages over the direct ignition. Where any chlorids of the alkalies are present in the ash there is danger of loss of potash from volatilization. This is avoided by the sulfate process. Moreover, there is not so much danger in this method of occluding particles of carbon in the ash.
234. The Destruction of Organic Matter by Moist Combustion.—In the process of ignition to destroy organic matter or remove ammonium salts in the determination of potash, there are often sources of error which may cause considerable loss. This loss, as has already been mentioned, may arise from the volatilization of the potash salts or mechanically from spattering. In order to avoid these causes of error de Roode has used aqua regia both for the destruction of the ammonium salts and for the oxidation of the organic matter at least sufficiently to prevent any subsequent reduction of the platinum chlorid.[193] His method consists in boiling a sample of the fertilizer, or an aliquot portion of a solution thereof with aqua regia. The proposed method has not yet had a sufficient experimental demonstration to warrant its use, but analysts may find it profitable to compare this process with the standard methods. The organic matter may also be destroyed by combustion with sulfuric acid, as in the kjeldahl method for nitrogen. The residue, however, contains ammonium sulfate and a large excess of sulfuric acid, and for both reasons would not be in a fit condition for the estimation of potash.
It is suggested that the organic matter might also be destroyed by boiling with strong hydrochloric acid, to which from time to time, small quantities of sodium chlorate free of potash is added. Subsequently the solution could be boiled with addition of a little nitric acid and the ammonium salts be removed.
235. Occurrence and History.—The generally accepted theory of the manner in which potash has been collected into deposits suited to use as a fertilizer has already been described.[194] The Stassfurt deposits, which have for many years been almost the sole source of potash in fertilizers, were first known as mines of rock salt. In 1839, having previously been acquired by the Prussian treasury, they were [Pg 257] abandoned by reason of the more economical working of rock salt quarries in other localities.[195] It was determined thereafter to explore the extent of these mines by boring, and a well was sunk to the depth of 246 meters, when the upper layer of the salt deposit was reached. The boring was continued into the salt to a total depth of 581 meters without reaching the bottom. The results of these experiments were totally unexpected. Instead of getting a brine saturated with common salt, one was obtained containing large quantities of potassium and magnesium chlorids.[196] Shafts were sunk in other places, and with such favorable results, that in 1862 potash salts became a regular article of commerce from that locality. At first these salts were regarded as troublesome impurities in the brine from which common salt was to be made, but at this time the common salt has come to be regarded as the disturbing factor. At the present time the entire product is controlled by a syndicate of nine large firms located at Stassfurt and vicinity. Outside of the syndicate properties a shaft has been sunk at Anderbeck, (Halberstadt,) which, however, has produced only carnallit, since kainit has not been found there. Also at Sondershausen, potash salts have been discovered and a shaft is now sinking there.
It is thus seen that the potash deposits extend over a wide area in Germany, and there is little fear of the deposits becoming exhausted in many centuries. In this country no potash deposits of any commercial importance have been discovered; but the geological conditions requisite to these formations have not been wanting, and their future discovery is not improbable. [Pg 258]
Figure. 17.
Geological Relations of the Potash
Deposits near Stassfurt.
236. Changes in Potash Salts in Situ.—The deposits of potash salts are not all found at the present in the same condition in which they were first deposited from the natural brines. The layers of salt have been subjected to the usual upheavals and subsidences peculiar to geological history. The layers of salt were thus tilted and the edges often brought to the surface. Here they were exposed to solution, and the dissolved brine afterward separated its crystallizable salts in new combinations. For instance, kieserit and the potassium chlorid of the carnallit were first dissolved and there was left a salt compound chiefly of potassium and sodium chlorids, sylvinit. In some cases there was a mutual reaction between the magnesium sulfate and the potassium chlorid and the magnesium potassium sulfate, schönit, was thus produced. This salt is also prepared at the mines artificially. The most important of these secondary products however, from the agricultural standpoint, is kainit. This salt arose by the bringing together of potassium sulfate, magnesium sulfate, and magnesium [Pg 259] chlorid, and was formed everywhere about the borders of the layers of carnallit wherever water could work upon them. In quantity the kainit, as might be supposed, is far less than the carnallit, the latter existing in immense deposits. There is however quite enough of it to satisfy all the demands of agriculture for an indefinite time. In fact for many purposes the carnallit can take the place of kainit without detriment to the growing crops. The relative positions and quantities of the layers of mineral matters in the potash mines, and the depth in meters at which they are found is shown in Fig. 17.[197]
237. Kainit.—The most important of the natural salts of potash for fertilizing purposes is the mixture known as kainit. It is composed in a pure state of a molecule each of potassium sulfate, magnesium sulfate, magnesium chlorid, and water. Chemically it is represented by the symbols:
K₂SO₄·MgSO₄·MgCl₂·H₂O. Its theoretical percentage of potash (K₂O), oxygen = 16, is 23.2.
Pure kainit, however, is never found in commerce. It is mixed naturally as it comes from the mines with common salt, potassium chlorid, gypsum, and other bodies. The content of potash in the commercial salt is therefore only a little more than half that of the pure mineral. In general it may be taken at 12.5 per cent, of which more than one per cent is derived from the potassium chlorid present. The following analysis given by Maercker may be regarded as typical:[198]
| Potassium | sulfate | 21.3 | per cent | |
| Magnesium | “ | 14.5 | “ | |
| Magnesium | chlorid | 12.4 | “ | |
| Potassium | “ | 2.0 | “ | |
| Sodium | “ | 34.6 | “ | |
| Calcium | sulfate | (gypsum) | 1.7 | “ |
| Water | 12.7 | “ | ||
| Alumina | 0.8 | “ | ||
Kainit occurs as a crystalline, partly colorless, partly yellow-red mass. When ground, in which state it is sent into commerce, it forms a fine, gray-colored mass containing many small yellow and red fragments. It is not hygroscopic and if it become moist it is due to the excess of common salt which it contains. [Pg 260]
According to Maercker kainit was formerly regarded as a potassium magnesium sulfate. But this conception does not even apply to the pure salt much less to that which comes from the mines. If, therefore, the agronomist desire a fertilizer free from chlorin he would be deceived in choosing kainit which may sometimes contain nearly fifty per cent of its weight of chlorids.
Where a fertilizer free of chlorin is desired, as for instance, in the culture of tobacco, kainit cannot be considered. In many other cases, however, the chlorin content of this body instead of being a detriment may prove positively advantageous, the chlorids on account of their easy diffusibility through the soil serving to distribute the other ingredients.
By reason of the presence of common salt and magnesium chlorid the ground kainit delivered to commerce tends to harden into compact masses. To prevent this in Germany it is recommended to mix it with about two and a half per cent of fine-ground dry peat.
Such a mixture is recommended in all cases where the freshly ground kainit is not to be immediately applied to the soil.
238. Carnallit.—This mineral is a mixture of even molecules of potassium and magnesium chlorids crystallized with six molecules of water. It is represented by the symbols KCl·MgCl₂·6H₂O. As it comes from the mines it contains small quantities of potassium and magnesium sulfates and small quantities of other accidental impurities. Existing as it does in immense quantities it has been extensively used for the manufacture of the commercial potassium chlorid (muriate of potash). For many purposes in agriculture, for instance, fertilizing tobacco fields, it is not suited, and it is less widely used as a fertilizer in general than its alteration product kainit. Its direct use as a fertilizer however is rapidly increasing since later experience has shown that chlorin compounds are capable of a far wider application in agriculture without danger of injury than was formerly supposed. [Pg 261] As it comes from the mines, the Stassfurt carnallit has the following composition:[199]
| Potassium | chlorid | 15.5 | per | cent. |
| Magnesium | “ | 21.5 | “ | “ |
| Magnesium | sulfate | 12.1 | “ | “ |
| Sodium | chlorid | 22.4 | “ | “ |
| Calcium | sulfate | 1.9 | “ | “ |
| Water | 26.1 | “ | “ | |
| Undetermined | 0.5 | “ | “ | |
Pure carnallit would have the following composition:
| Chlorin | 38.3 | per | cent. |
| Potassium | 14.0 | “ | “ |
| Magnesium | 8.7 | “ | “ |
| Water | 39.0 | “ | “ |
Equivalent to
| Potassium | chlorid | 26.8 | “ | “ |
| Magnesium | “ | 34.2 | “ | “ |
| Water | 39.0 | “ | “ | |
The commercial article as taken from the mines, as is seen above, has less potash (K₂O) than kainit, the mean content being about nine and nine-tenths per cent. Those proposing to use this body for fertilizing purposes should bear in mind that it contains less potash and more chlorin than kainit.
Carnallit occurs in characteristic brown-red masses. On account of its highly hygroscopic nature it should be kept as much as possible out of contact with moist air and should not be ground until immediately before using.
By reason of the greater bulk in proportion to its content of potash and its hygroscopic nature and consequent increased difficulty in handling, the price per unit of potash in carnallit is less than in kainit.
In some localities small quantities of ammonium chlorid have been found with carnallit but not to exceed one-tenth per cent. It has therefore no practical significance to the farmer but may be of interest to the analyst.
239. Polyhalit.—Polyhalit is a mineral occurring in Stassfurt deposits and consisting of a mixture of potassium, magnesium, and calcium sulfates, with a small proportion of crystal water. This mineral, on account of its being practically free of chlorin, would be one especially desirable for use in those cases, as in the culture of tobacco, where chlorids are injurious. Unfortunately, it does not [Pg 262] occur in sufficient quantities to warrant the expectation of its ever being found in masses large enough to become a general article of commerce. It is found only in pockets or seams among the other Stassfurt deposits, and there is no assurance given on finding one of these deposits of polyhalit that it will extend to any great distance. The composition of the mineral is shown by the following formula: K₂SO₄·MgSO₄·(CaSO₄)₂·H₂O. Its percentage composition is shown by the following numbers:
| Potassium sulfate | 28.90 | per | cent. |
| Magnesium sulfate | 19.93 | “ | “ |
| Calcium sulfate | 45.18 | “ | “ |
| Water | 5.99 | “ | “ |
The percentage of potash corresponding to the above formula is 15.62. It therefore contains a considerable excess of potash over kainit, and on account of its freedom from chlorids, would be preferred for many purposes.
240. Krugit.—This mineral occurs associated with polyhalit and differs from it only in containing four molecules of calcium sulfate instead of two. Its formula is: K₂SO₄·MgSO₄·(CaSO₄)₄·H₂O. As it comes from the mines it is frequently mixed with a little common salt. Its mean percentage composition as it comes from the mines is given in the following numbers:
| Potassium sulfate | 18.60 | per | cent. |
| Magnesium sulfate | 14.70 | “ | “ |
| Calcium sulfate | 61.00 | “ | “ |
| Sodium chlorid | 1.50 | “ | “ |
| Water | 4.20 | “ | “ |
The percentage of potash corresponding to the above formula is 10.05. It is therefore less valuable than kainit in so far as its content of potash is concerned. This salt also exists in limited quantities and is not likely to become an important article of commerce.
241. Sylvin.—One of the alteration products of carnallit is a practically pure potassium chlorid which, as it occurs in the Stassfurt mines is known as sylvin. The alteration of the carnallit arises from its solution in water from which, on subsequent evaporation, the potassium chlorid is deposited alone. This mineral is found in only [Pg 263] limited quantities in the Stassfurt deposits and it therefore does not have any great commercial importance.
242. Sylvinit.—This mineral has been mined in recent years in considerable quantities. It is, in fact, only common salt carrying large quantities of potassium chlorid together with certain other accidental impurities. It was probably formed by the drying up of a saline mass in such a way as not to permit the complete separation of its mineral constituents. The average composition of sylvinit as it comes from the mines is given in the following table:
| Potassium chlorid | 30.55 | per cent. |
| Sodium chlorid | 46.05 | “ |
| Potassium sulfate | 6.95 | “ |
| Magnesium sulfate | 4.80 | “ |
| Magnesium chlorid | 2.54 | “ |
| Calcium sulfate | 1.80 | “ |
| Water and insoluble | 7.29 | “ |
This salt is richer in chlorin than any other of the Stassfurt potash minerals, containing altogether 79.14 per cent of chlorids. Its potash content amounts to 23.04 per cent, but in proportion to the potash which it contains, it is relatively poorer in chlorin than kainit and carnallit. On account of its high content of potash the freights on a given weight thereof as contained in sylvanit are lower than for kainit and carnallit.
243. Kieserit.—The mineral kieserit is essentially magnesium sulfate and it does not necessarily contain any potash salts. Under the name of kieserit, however, or bergkieserit, there is mined a mixture of carnallit and kieserit, which is a commercial source of potash. The mixture contains the following average content of the bodies named:
| Potassium chlorid | 11.80 | per cent. |
| Magnesium sulfate | 21.50 | “ |
| Magnesium chlorid | 17.20 | “ |
| Sodium chlorid | 26.70 | “ |
| Calcium sulfate | 0.80 | “ |
| Water | 20.70 | “ |
| Insoluble | 1.30 | “ |
This mixture contains only about seven per cent of potash and would not prove profitable when used at a distance from the mines on account of the cost of freights. It has proved valuable, however, for a top [Pg 264] dressing for meadow lands in the vicinity of Stassfurt.
244. Schönit.—Among the Stassfurt deposits there occurs in small quantities a mineral, schönit, which is composed of the sulfates of potassium and magnesium. The quantity of the mineral occurring naturally is very small and therefore it has no commercial importance. When, however, kainit is washed with water the common salt and magnesium chlorid which it contains being more soluble are the first leached out, and the residue has approximately the composition of the pure mineral. This mixture, as prepared in the way mentioned above, has the following average composition:
| Potassium sulfate | 50.40 | per cent. |
| Magnesium sulfate | 34.00 | “ |
| Sodium chlorid | 2.50 | “ |
| Water | 11.60 | “ |
The percentage of potash corresponding to the above composition is 27.2. This substance being so rich in potash, and practically free of chlorids, is well suited to transportation to great distances and for general use in the field. Since, however, a considerable expense attends the manufacture of the artificial schönit, the advantages above named give it very little, if any, advantage in competition with the other potash salts as they come from the mines. It has, however, an especial value for the fertilization of tobacco and vineyards.
245. Potassium Sulfate.—Several grades of potassium sulfate are found in the market for fertilizing purposes, some of them quite pure, containing over ninety-seven per cent of the pure sulfate. The following data show the composition of a high grade and low grade potassium sulfate of commerce:
| High grade. | Low grade. | |||
| Potassium sulfate | 97.20 | per cent. | 90.60 | per cent. |
| Potassium chlorid | 0.30 | “ | 1.60 | “ |
| Magnesium sulfate | 0.70 | “ | 2.70 | “ |
| Magnesium chlorid | 0.40 | “ | 1.00 | “ |
| Sodium chlorid | 0.20 | “ | 1.20 | “ |
| Insoluble | 0.20 | “ | 0.30 | “ |
| Water | 0.70 | “ | 2.20 | “ |
Naturally, high grade sulfates of this kind can only be prepared in chemical factories built especially for the work. The result is that [Pg 265] the potash per unit is raised greatly in price. When, however, the fertilizers are to be transported to a great distance, the saving in freight often more than compensates for the higher price of the potash. It therefore happens that there are many places in this country where the actual price of potash per pound is less in high grade sulfates than in kainit or carnallit. When, in addition to this, the especial fitness of the high grade sulfates for certain forms of fertilization, especially tobacco growing, is considered, it is seen that at this distance from the mines these high grade salts are of no inconsiderable importance. The percentage of potash in the high grade sulfates often exceeds fifty.
246. Potassium Magnesium Carbonate.—This salt has lately been manufactured and used to a considerable extent, especially for tobacco fertilizing. As furnished to the trade it has the following average composition:
| Potassium carbonate | 35 to 40 | per cent. |
| Magnesium carbonate | 33 to 36 | “ |
| Water of crystallization | 25 | “ |
| Potassium chlorid, potassium sulfate, | ||
| and insoluble | 2 to 3 | “ |
The content of potash, as is seen from the above formula, amounts to from seventeen to eighteen per cent. The compound is completely dry, is not hygroscopic, and is, therefore, always ready for distribution. It is especially to be recommended for all those intensive cultures where it is feared that chlorids and sulfates will prove injurious, especially in the cultivation of tobacco.
247. Potash in Factory Residues.—The residues from the potash factories in Stassfurt and vicinity contain considerable quantities of potash and attempts have been made to recover this waste and put it into form for fertilizing uses. The waste waters of the factories are sometimes collected and evaporated, and the residue incinerated. The content of potash in these residues is extremely variable, usually quite low, and they, therefore, cannot be recommended for fertilizing purposes, especially if they are to be transported to any distance.
[Pg 266] 248. Quantity of Potash Salts Used.—The total quantity of potash delivered to consumers from the Stassfurt mines in 1891, the last year for which complete statistics are at hand was 413,508 tons of kainit and sylvinit, 39,444 tons of carnallit, 18,078 tons of sulfate, and 12,453 tons of the potassium magnesium sulfate. Of the above quantities, 115,245 tons of kainit were shipped to North America, and of the high grade sulfate mentioned, 13,322 tons were sent to other countries, and of the potassium magnesium sulfate, 11,081 tons were exported.
249. Classification of Methods.—To detect the presence of potash in a mixture the aid of the spectroscope may be invoked. In the scale of the spectrum divided into 170 parts, on which the sodium line falls at 50, potassium gives three faint rather broad bands, two red, falling at 17 and 27, and one plum-colored band, near the extreme right of the spectrum, at 153. Potassium, however, does not give brilliant and well-marked spectral bands, such as are afforded by its associates rubidium, caesium, sodium, and lithium. A convenient qualitative test which, for practical purposes will be quite sufficient, may be secured by dipping a platinum loop into a strong acid solution of the supposed potash compound, and viewing through a piece of cobalt glass, the coloration produced thereby when held in the flame of a bunsen. The red-purple tint thereby produced should be compared with that coming from a pure potash salt similarly treated. If a fertilizer sample give no indication of potash when treated as above it may be safely concluded that it does not contain any weighable quantity of potash.
For the estimation of the percentage of potash present in a given sample it may be safely assumed that all of value in agriculture will be given up to an aqueous or slightly acid solution if organic matter have been destroyed as indicated in a previous paragraph. In the case of minerals insoluble in a dilute acid the potash may be determined by some one of the processes given in the first volume.[200] The potash having been obtained in an aqueous or slightly acid (hydrochloric) solution, it may be determined either by precipitation as potassium platinochlorid or as potassium perchlorate. The former method is the one [Pg 267] which has been almost exclusively used by analysis in the past, but the latter one is coming into prominence and by reason of the greater economy attending its practice and the excellent results obtained by some analysts, demands a generous consideration.
250. The Platinic Chlorid Method.—The principle of this method rests on the great insolubility of the potassium platinochlorid in strong alcohol and the easy solubility of some of its commonly attending salts; viz., sodium, etc., in the same reagent. Before the precipitation of the potash it is necessary to remove the bases of the earths, sulfates, etc. Barium chlorid and hydroxid, ammonium oxalate or carbonate, sulfuric acid, etc., are used in conjunction or successively to effect these purposes in the manner hereinafter described. The filtrate and washings containing the potash are evaporated to dryness and gently ignited to expel excess of ammonium salts and in the residue taken up with water and acidulated with hydrochloric acid, the potash is precipitated with platinic chlorid solution. The best methods of executing the analysis follow.
251. The Official Agricultural Method.—This method is based on the processes at first proposed by Lindo[201] and Gladding,[202] and is given below as adapted to mixed fertilizers and mineral potash salts.[203]
(1) In Superphosphates.—Boil ten grams with 300 cubic centimeters of water thirty minutes. To the hot solution add ammonia in slight excess, and then a sufficient quantity of ammonium oxalate to precipitate all the lime present; cool and make up to half a liter, mix thoroughly, and filter through a dry filter; evaporate fifty cubic centimeters, corresponding to one gram, nearly to dryness, add one cubic centimeter of dilute sulfuric acid (1 to 1), evaporate to dryness and ignite to whiteness. As all the potash is in form of sulfate, no loss need be apprehended by volatilization of potash, and a full red heat must be maintained until the residue is perfectly white. This residue is dissolved in hot water, plus a few drops of hydrochloric acid, and a slight excess of platinum solution is added. This solution is then evaporated to a thick paste in a small dish, and eighty per cent alcohol added. In evaporating, special precaution should be taken [Pg 268] to prevent absorption of ammonia. The precipitate is washed thoroughly with alcohol by decantation and on the filter, as usual. The washing should be continued even after the filtrate is colorless. Ten cubic centimeters of the ammonium chlorid solution, prepared as hereinafter directed, are run through the filter, or the washing may be performed in the dish. The ten cubic centimeters will contain the bulk of the impurities, and are thrown away. Fresh portions of ten cubic centimeters of the ammonium chlorid are run through the filter several times (5 or 6). The filter is then washed thoroughly with pure alcohol, dried, and weighed as usual. Care should be taken that the precipitate is perfectly soluble in water. The platinum solution used contains one gram of metallic platinum in every ten cubic centimeters. To prepare the washing solution of ammonium chlorid, place in a bottle 500 cubic centimeters of water and 100 grams of ammonium chlorid and shake till dissolved. Now pulverize five or ten grains of potassium platinochlorid, put in the bottle and shake at intervals for six or eight hours; let settle over night, then filter off the liquid into a second bottle. The first bottle is then ready for preparation of a fresh supply when needed.
(2) Potassium Chlorids.—In the analysis of these salts an aliquot portion of the solution, containing a half gram, is evaporated with forty cubic centimeters of the platinum solution and a few drops of hydrochloric acid, and washed as before.
(3) Potassium Sulfate, Kainit, Etc.—In the analysis of kainit, dissolve ten grams of the pulverized salt in 300 cubic centimeters of boiling water, add ammonia to slight excess, then a sufficient quantity of ammonium oxalate to throw down all lime present; cool and make up to half a liter, mix thoroughly, and filter on a dry filter; from twenty-five cubic centimeters, corresponding to a half gram, proceed to remove the ammonia, as in the analysis of superphosphates; dissolve the residue in hot water, plus a few drops of hydrochloric acid, and add fifteen cubic centimeters of platinum solution. In the analysis of high-grade sulfate and of double-manure salt (potassium sulfate, magnesium sulfate, containing about twenty-seven per cent of potassium oxid), make up the solution as above, but omit the precipitation, evaporation, etc.; to an aliquot part equal to a half gram add fifteen [Pg 269] cubic centimeters of platinum solution. In all cases special care must be taken in the washing with alcohol to remove all the double platinum sodium chlorid which may be present. The washing should be continued some time after the filtrate is colorless. Twenty-five cubic centimeters of the ammonium chlorid solution are employed instead of ten cubic centimeters, and the twenty-five cubic centimeters poured through at least six times to remove all sulfates and chlorids. Wash finally with alcohol; dry and weigh as usual.
252. Alternate Method for Potash.—Boil ten grams of the prepared sample for thirty minutes with 300 cubic centimeters of water, and, after cooling and without filtering, make up to one liter and filter through a dry filter. If the sample have ten per cent of potassium oxid, use fifty cubic centimeters of the filtrate; if less than ten per cent of potassium oxid (ordinary potash fertilizers), use 100 cubic centimeters of the filtrate. In each case make the volume up to 150 cubic centimeters, heat to 100°, and add, drop by drop with constant stirring, a slight excess of barium chlorid, and, without filtering, in the same manner add barium hydrate in slight excess. Filter while hot and wash until the precipitate is free of chlorids. Add to the filtrate one cubic centimeter of strong ammonium hydrate, and then a saturated solution of ammonium carbonate, until the excess of barium is precipitated. Heat and add, in fine powder, a half gram of pure oxalic acid or 0.75 gram of ammonium oxalate. Filter, wash free of chlorids, evaporate the filtrate to dryness in a platinum dish, and ignite carefully over the free flame, below red heat, until all volatile matter is driven off.
The residue is digested with hot water, filtered through a small filter, and washed with successive small portions of water until the filtrate amounts to thirty cubic centimeters or more. To this filtrate, add two drops of hydrochloric acid, in a porcelain dish, and from five to ten cubic centimeters of a solution of ten grams of platinic chlorid in 100 cubic centimeters of water. The mixture is evaporated on a water-bath to a thick sirup, as above, treated with alcohol of eighty per cent strength, washed by decantation, collected in a gooch or other form of filter, washed with strong alcohol, afterwards with five cubic [Pg 270] centimeters of ether, dried for thirty minutes at 100°, and weighed.
It is desirable, if there be an appearance of foreign matter in the double salt, that it should be washed, according to the previous method, with ten cubic centimeters of the half-concentrated solution of ammonium chlorid, which has been saturated by shaking with potassium platinochlorid.
253. Method of Solution for Organic Compounds.—In case the potash is contained in organic compounds, like tobacco stems, cottonseed hulls, etc., weigh ten grams, saturate with strong sulfuric acid, and ignite in a muffle to destroy organic matter. Add a little strong hydrochloric acid to moisten the mass and warm slightly so as to loosen it in the dish. Proceed then as in the lindo-gladding or alternate method.
254. Factors.—The use of the factors 0.3056 for converting potassium platinochlorid to potassium chlorid and 0.19308 for converting it to potassium oxid is advised. The latter number is almost identical with that used by the Halle and Stassfurt chemists viz., 0.1927 and 0.1928 respectively.
255. Methods Used at the Halle Station.—(1) In Kainits and other Mineral Salts of Potash.[204]—Five grams of the prepared sample are boiled for half an hour in a half liter flask with from twenty to thirty cubic centimeters of concentrated hydrochloric acid and 100 cubic centimeters of water, and afterwards as much water added as is necessary to fill the flask about three quarters full, and the sulfuric acid is then precipitated with barium chlorid. To avoid an excess of barium chlorid the solution used is of known strength and is added first in such quantity as would precipitate the sulfuric acid from a kainit of low sulfuric acid content. The mixture is then boiled, allowed to settle and tried with a dropping tube containing barium chlorid. If a further precipitate be given a few drops more of barium chlorid solution are added, again boiled and allowed to settle. This is continued until barium chlorid gives no precipitation. After the barium chlorid gives no more precipitate a drop of dilute sulfuric acid is added to test for excess of barium. The operation is continued with the sulfuric acid until it no longer gives a precipitate of barium sulfate. By the alternate [Pg 271] use of the barium chlorid and sulfuric acid the exact neutral point can soon be secured. When this point is reached the liquid is allowed to cool, the flask is filled to the mark, its contents filtered, and of the filtrate fifty cubic centimeters, equal to half a gram of the substance, taken for further estimation.
This quantity is evaporated on a water-bath to a sirupy consistence in a porcelain dish with ten cubic centimeters of platinic chlorid. The platinic chlorid solution should contain one gram of platinum in each ten cubic centimeters. The residue is treated with eighty per cent alcohol and, with stirring, allowed to stand for an hour. The precipitate is then collected on a gooch, either of platinum or porcelain, washed about eight times with eighty per cent alcohol and the potassium platinochlorid dried for two hours at 100°. After weighing the precipitate is dissolved in hot water and the residue washed under pressure, first with hot water and then with alcohol. The crucible with the asbestos felt is dried at 100° and weighed. Any impurities which the double salt may have carried down with it are left on the filter and the weight of the original precipitate can thus be corrected. The weight of potassium platinochlorid is multiplied by 0.1927 and the product corresponds to the weight of K₂O in the sample taken.
(2) Estimation of Potash in Guanos and Other Fertilizers containing Organic Substances.—Ten grams of the substances are carefully incinerated at a low temperature in a platinum dish. After ignition the contents of the dish are placed in a half liter flask and boiled for an hour with hydrochloric acid and a few drops of nitric acid. The sulfuric acid can then be precipitated directly with barium chlorid, or better, allow the flask to cool, fill to the mark, filter and treat an aliquot part of the filtrate with barium chlorid as described above. The filtrate from the separated sulfate of barium is neutralized with ammonia and all the bases, with the exception of magnesia and the alkalies, precipitated with ammonium carbonate; boil, fill to the mark and filter. Of this filtrate evaporate from 100 to 200 cubic centimeters in a platinum dish. After evaporation the ammonium salts are driven off by careful ignition, the residue taken up with hot water and filtered through as small a filter as possible into a porcelain [Pg 272] dish; the magnesia remaining in the precipitate. The filtrate is acidified with a few drops of hydrochloric acid, ten cubic centimeters of platinic chlorid added and the further determination conducted as with kainit.
256. Dutch Method.—The process used at the Royal Agricultural Station of Holland is almost identical with that employed at Halle.[205]
A. Method for Stassfurt and other Potash Salts.—The necessary reagents are:
1. A dilute solution of barium chlorid:
2. A solution of platinic chlorid containing one gram of platinum in ten cubic centimeters: It must be wholly free from platinous chlorid and nitric acid, and partially freed from an excess of hydrochloric acid by repeated evaporations with water.
3. Alcohol of eighty per cent strength by the volume:
The methods of bringing the potash into solution and of precipitating the sulfuric acid are the same as for the Halle process described above.
Add then twenty cubic centimeters of the platinum solution and evaporate the mixture nearly to dryness. Add a sufficient quantity of eighty per cent alcohol and stir for some time. Allow to stand and then filter through a gooch dried at 120°. Finally wash with eighty per cent alcohol, dry at 120°, and weigh.
B. Method for Potash Superphosphate and other mixed Fertilizers.—The reagents necessary are the same as under A, and, in addition, a saturated solution of barium hydrate and a solution of ammonium carbonate mixed with ammonia.
Boil twenty grams of the substance with water for half an hour, cool, make up to half a liter and filter. Boil fifty cubic centimeters of the filtrate, and add barium chlorid till no more precipitate forms. Mix with baryta water to strong alkaline reaction, cool, make up to 100 cubic centimeters and filter. Raise fifty cubic centimeters of the filtrate to the boiling temperature and add ammonium carbonate solution till no more precipitate forms: Cool, make up to 100 cubic centimeters and filter. Transfer fifty cubic centimeters of the filtrate to a platinum dish, evaporate and heat the residue, avoiding too high a [Pg 273] temperature, till the ammonia salts are expelled. Dissolve the residue in water, filter, and treat the filtrate as described under A.
257. Swedish Methods.—The Swedish chemists determine the potash in mineral salts by the platinum chlorid process, but with certain variations from the processes already given. The manipulation is conducted as follows:[206]
Weigh one gram of the sample to be examined and pour about 300 cubic centimeters of hot water over it in a beaker and filter after complete solution; add one cubic centimeter of hydrochloric acid, heat nearly to boiling, add dilute barium chlorid solution from a pipette or burette in a very fine stream stirring, slowly and carefully, till all sulfuric acid is completely precipitated, and only a trace of the precipitant is in excess. If the precipitation be conducted in the way given the barium sulfate will come down in crystalline condition, and settle rapidly within a few minutes, and almost immediately after the precipitation is finished may be filtered clear. The filtrate and washings from the barium sulfate are brought into a liter flask; fill this to the mark, take out fifty cubic centimeters with a pipette, evaporate the greater portion on a water-bath in a porcelain dish, transfer the residue by means of ammonia-free water to a beaker of fifty cubic centimeters capacity, add ten cubic centimeters of platinic chlorid solution, stir well with a glass rod, evaporate on a water-bath to a sirupy condition, allow to cool, and if the residue be too dry, add a few drops of water to allow the sodium platinochlorid to take up crystal water with certainty, stir well, add alcohol after a few minutes, mix carefully, leave the mixture standing for a while in the beaker covered with a watch glass, stirring occasionally; finally decant the solution, which must be of a dark yellow color, through a very small filter, wash the precipitate in the beaker repeatedly with small quantities of alcohol and decant; then transfer the precipitate to the filter, wash with alcohol, dry the filter and the precipitate at a gentle heat till all alcohol has evaporated, carefully transfer the contents of the filter to a watch glass placed on white glazed paper; dissolve the potassium platinochlorid still remaining on the filter in small quantities of boiling water, evaporate the filtrate on a water-bath in an accurately weighed platinum dish to dryness [Pg 274] and transfer the same to the main portion of the chlorid from the watch glass. In order to obtain the salt free of the corresponding combinations of sodium, barium, calcium, and magnesium, which salts, although soluble in alcohol, may make the salt impure, before weighing, treat the precipitate twice with small quantities of cold water which will dissolve these impurities; evaporate the solution after addition of one cubic centimeter of platinic chlorid nearly to dryness on a water-bath, treat the residue in the same way as given before, add the small quantity of potassium platinochlorid which is hereby obtained together with the main portion to the platinum dish, dry at 130°, and weigh. Only after having been treated in this way may the precipitated potassium platinochlorid be considered absolutely pure. The Stassfurt salts contain magnesia, often in large quantities and as a consequence the potassium platinochlorid precipitated directly is likely to be contaminated therewith.
258. Methods for the Analysis of Carnallit, Kainit, Sylvinit, and Kieserit.—The chemists of the German Potash Syndicate use the following methods in the analysis of the raw products mentioned above.[207]
(1) Preparation of the Sample.—It is advisable to take from a large well mixed mass at least half a kilogram for the analytical sample and this should be ground to a fine powder in a mill or mortar.
(2) Estimation of the Potash by the Precipitation Method.—In a half liter flask are placed 35.70 grams of kainit or sylvinit, or 30.56 grams of carnallit or bergkieserit, which are boiled with 350 cubic centimeters of water after the addition of ten cubic centimeters of hydrochloric acid. After cooling the flask is filled to the mark with water, well shaken, and its contents filtered. Fifty cubic centimeters of the filtrate are treated in a 200 cubic centimeter flask with a solution of barium chlorid, the flask filled to the mark, well shaken, and its contents filtered. Twenty cubic centimeters of the filtrate, corresponding to 0.3570 or 0.3056 gram of the substance, are treated with five cubic centimeters of platinic chlorid solution and the potassium estimated according to the usual methods. [Pg 275]
(3) Estimation of Potash (K₂O) in Raw Potash Salts.—(a) For the determination of potash alone in carnallit, kainit, and sylvinit one hundred grams of the well-mixed sample are put into a graduated flask holding one liter and dissolved by boiling with half a liter of water, acidulated with ten cubic centimeters of hydrochloric. The purpose of adding hydrochloric acid is to bring any polyhalit that might be present in the salts into solution and which it is difficult to dissolve in pure water. After dissolving and cooling the flask is filled up to the mark. The solution, after mixing, is filtered through a dry filter and 100 cubic centimeters of the filtrate, corresponding to ten grams substance, are put into a half liter flask by means of a pipette. After the addition of 200-300 cubic centimeters of water the solution is heated to boiling and the sulfuric acid accurately precipitated with normal barium chlorid solution, containing 104 grams of the dry salt in one liter. The volume of the precipitate is calculated from the amount of barium solution used and from the specific gravity of the barium sulfate. After cooling, the flask is filled up with water as far above the mark as equals the volume of the calculated barium precipitate, and, after thorough mixing, the solution is filtered again through a dry filter. Fifty cubic centimeters of this filtrate, corresponding to one gram substance, are evaporated upon the water-bath with a sufficient amount of platinic chlorid. The residue of potassium platinochlorid is washed with ninety per cent alcohol, dried at 120°, and weighed.
(b) If it be desired to determine separately the quantity of potash present in the form of sulfate and in the form of chlorid, as for example in kainit and in sulfate of potash, or if it is to be determined whether potassium sulfate is in combination with a proportionate amount of magnesium chlorid, as in kainit, or in combination with magnesium sulfate alone, as in schönit, it then becomes necessary to determine besides potash the percentages of chlorin, sulfuric acid, lime, magnesia, the total alkalies, water, and the residue insoluble in water. For this purpose 100 grams of the sample are dissolved, the solution is filtered, the filter washed, and the filtrate made up to one liter; a part of the liquid is taken for the determination of sulfuric acid; by precipitating with barium [Pg 276] chlorid, and another part for the determination of lime and magnesia. For the determination of the alkali chlorids, 100 cubic centimeters of the solution, corresponding to ten grams substance, are acidulated with hydrochloric, and, after heating to boiling, the sulfuric acid is completely precipitated with barium chlorid, with the precaution of using not more of the barium solution than is necessary for the complete precipitation. Fifty cubic centimeters of the filtered solution, corresponding to one gram substance, are evaporated to dryness in order to drive off the hydrochloric acid. Magnesium chlorid is decomposed by igniting with oxalic acid or with mercuric oxid. After ignition, the residue is moistened with a little ammonium carbonate for the purpose of converting the calcium oxid that may have been formed into calcium carbonate. The alkali chlorids, which are entirely free of lime and magnesia, are weighed, and potassium chlorid is determined by means of platinic chlorid. The amount of sodium chlorid is obtained by deducting potassium chlorid from the mixed chlorids. For the water determination five grams of the sample are ignited and the loss of weight is determined. The ignited mass is dissolved in water, and for the purpose of determining the quantity of magnesium chlorid that may have been decomposed by the ignition the percentage of chlorin is determined by titration. The difference in the contents of chlorin before and after ignition is subtracted from the loss in weight, after allowance has been made for the absorption of oxygen and for the loss of hydrogen. The rest is water. The results obtained are calculated in the following manner: From the total amount of the sulfuric acid found, that portion is deducted which is combined with calcium as calcium sulfate; the rest of the sulfuric acid is divided into two equal parts for the purpose of calculating the contents of potassium sulfate and magnesium sulfate, according to the molecular proportion in which these salts are present in kainit and in schönit. If there be an excess of potash left uncombined with sulfuric acid, then it is in the form of potassium chlorid; likewise the amount of magnesia, uncombined with sulfuric acid, is to be reckoned as magnesium chlorid. The result of this calculation will tell how much potash is in the form of kainit [Pg 277] (K₂SO₄, MgSO₄, MgCl₂ with 6H₂O) and how much of it is in the form of schönit (K₂SO₄, MgSO₄, with 6H₂O) and how much in the form of potassium chlorid. The sodium is reckoned as sodium chlorid.
(c) In calculating the contents of potash, of potassium chlorid, and of potassium sulfate from the weighed potassium platinochlorid, the factors 0.1928, 0.3056, and 0.3566 are used, assuming that the atomic weight of platinum is 197.18.
(d) The two methods which have been described under a and b, and which are in common use in the Stassfurt potash industry, i. e., the so-called precipitation method, and the oxalic acid method, give almost identical results. The first method, however, deserves preference on account of greater simplicity in cases where potash alone is to be determined. Finkner’s method likewise gives results which agree well with the results obtained by the customary methods. It consists in evaporating the salt solution with a sufficient quantity of platinic chlorid without previously removing the sulfuric acid, reducing the potassium platinochlorid, and weighing the metallic platinum.
The following are the results of comparative analyses:
| 1. After the precipitation method | 22.02 per cent KCl |
| 2. After the oxalic acid method | 22.03 per cent KCl |
| 3. After Finkner’s method | 22.01 per cent KCl |
In another sample of carnallit the following results were obtained:
| 1. After the precipitation method | 17.88 per cent KCl |
| 2. After the oxalic acid method | 17.88 per cent KCl |
In a third sample of carnallit the content of potassium chlorid was as follows:
| 1. After the precipitation method | 18.44 per cent KCl |
| 2. After the oxalic acid method | 18.38 per cent KCl |
The Anhalt chemists object to precipitating the sulfuric acid and alkaline earths with barium oxid and ammonium carbonate, and afterwards the potash with platinic chlorid. The results obtained with this method are, according to them, very inaccurate, and always too low. This is explained by the fact that it is impossible to precipitate sulfuric acid without at the same time precipitating some of the potash, unless it be in an acid solution. [Pg 278]
A separation of the alkaline earths, if potash alone is to be determined, is superfluous, for the reason that calcium and magnesium platinochlorid are soluble in ninety per cent alcohol, even with more facility than sodium platinochlorid.
259. Methods for Concentrated Potash Salts.—In the preceding paragraphs have been given the methods used by the Stassfurt syndicate for the estimation of potash in the raw salts as they come from the mines. Following are the methods used by the same syndicate for the concentrated approximately pure compounds and the other salts which accompany them.
Potassium Chlorid.—The following process is used for the estimation of potassium and other constituents of the high grade chlorids of commerce. In a half liter flask are placed 7.6405 grams of the finely powdered sample, which is dissolved and made up to the mark. With salts which contain more than half a per cent of sulfuric acid the preliminary conversion of the sulfates into the corresponding chlorin compounds, by precipitation with barium chlorid solution, is necessary. Twenty cubic centimeters of the above solution, corresponding to 0.3056 gram of the salt, are placed in a flat porcelain dish having a diameter of about ten centimeters and, after the addition of five cubic centimeters of the platinic chlorid solution, evaporated on the water-bath with constant stirring until, after cooling, the sirupy liquid passes quickly into a fine crystalline condition. The residue is rubbed into a fine powder with a glass rod, mixed with twenty cubic centimeters of ninety-six per cent alcohol and dried at 120° to a constant weight. It is weighed while warm and brought on a moistened filter with alcohol, care being taken that the liquid does not touch the edge of the filter. The filtration can be carried on under a moderate pressure. The complete washing of the potassium platinochlorid can be easily accomplished upon the filter. The filter and the precipitate, after as much of the alcohol wash has been removed as is possible, are dried at 120° to constant weight and weighed while still warm. One milligram of the potassium platinochlorid thus obtained corresponds to a tenth per cent of potassium chlorid.
Estimation of Sodium Chlorid.—For the estimation of the sodium chlorid which may be present in the potassium chlorid twelve and a half [Pg 279] grams of the latter salt are dissolved in a quarter liter flask with twenty-five cubic centimeters of boiling water after the addition of a little potassium carbonate for the purpose of converting the magnesium and calcium compounds into carbonates. After filtration 100 cubic centimeters corresponding to five grams of the salt are evaporated to dryness in a porcelain or platinum dish after the addition of a few drops of concentrated hydrochloric acid in order to convert any calcium carbonate which may be present into chlorid. The residue is gently ignited and weighed. In this mixture of potassium and sodium chlorids the potassium chlorid may be estimated in the usual way and the sodium chlorid determined by difference or the respective proportions of the two bases may be calculated after the determination of the total chlorin by precipitation with a standard solution of silver nitrate.
Estimation of Magnesium Chlorid.—In order to estimate the amount of magnesium chlorid in high grade muriate of potash, twenty-five grams of the latter salt are dissolved in a half liter flask and treated with ten cubic centimeters of a normal solution of potash lye. The flask is then filled to the mark with water, thoroughly shaken and its contents filtered. Fifty cubic centimeters of the filtrate are then titrated with one-tenth normal sulfuric acid. The calcium compounds which remain in solution do not influence the result. The quantity of magnesium chlorid originally present corresponds to the number of cubic centimeters of the normal potash lye which has disappeared in the operation. The reaction which takes place is represented by the following equation:
MgCl₂ + 2KOH = MgO₂H₂ + 2KCl.
Potassium Sulfate.—The quantity of potassium sulfate contained in the high grade sulfates of commerce is determined in the following manner: In a half liter flask are placed 8.9235 grams of the finely ground sample which is dissolved in about 350 cubic centimeters of boiling water after the addition of twenty cubic centimeters of hydrochloric acid. The sulfuric acid is thrown out by the addition, drop by drop, of a barium chlorid solution, the contents of the flask being kept boiling meanwhile and thoroughly stirred. From time to time the addition of the barium chlorid is stopped and the upper part of the liquid allowed to become clear by the subsidence of the barium sulfate. [Pg 280] It is then noticed whether or not an additional drop of the barium chlorid solution produces a turbidity. Any excess of barium chlorid is removed by the careful addition of sulfuric acid. After the precipitation is complete and the contents of the flask are cooled, it is filled up to the mark with water and its contents filtered. Twenty cubic centimeters of the filtrate, corresponding to 0.357 gram of the original salt are precipitated by platinic chlorid in the usual manner and the resulting potassium platinochlorid collected and weighed. One milligram of the potassium platinochlorid thus obtained corresponds to one-tenth per cent of potassium sulfate in the original salt. To the percentage of potassium sulfate thus found three-tenths per cent are to be added for a correction when high grade potassium sulfate is taken. If the sample be a high grade sulfate of potassium and magnesium no correction should be applied.
Estimation of Potassium Chlorid and Potassium Sulfate in Calcined Manurial Salts.—In these salts 15.281 grams for potassium chlorid or 17.847 grams for potassium sulfate are dissolved in a half liter flask after the addition of ten cubic centimeters of hydrochloric acid. The flask is filled to the mark and its contents filtered and 250 cubic centimeters placed in a half liter flask and treated with barium chlorid solution as indicated above. The rest of the operation is exactly as has been described. In each case one milligram of the potassium platinochlorid corresponds to one-tenth per cent of the desired salt.
Estimation of Magnesium Sulfate in Kieserit.—Ten grams of the finely powdered kieserit are boiled for one hour in a half liter flask two-thirds full of water. After cooling, from fifty to sixty cubic centimeters of double normal potash lye and twenty cubic centimeters of a ten per cent neutral potassium oxalate solution are added, the flask filled to the mark, and after being well shaken and standing for a quarter of an hour, filtered. The reaction is represented by the formula
MgSO₄ + 2KOH = MgO₂H₂ + K₂SO₄.
Fifty cubic centimeters of the filtrate are then titrated with one-tenth normal sulfuric acid. To the percentage of magnesium sulfate found by this process two-tenths per cent are to be added as a correction.
Barium Chlorid Solution.—Dissolve 122 grams of crystallized barium chlorid in water in a liter flask. Add fifty cubic centimeters of [Pg 281] hydrochloric acid and water to the mark and shake well.
260. The Barium Oxalate Method.—The principle of this process, worked out by Schweitzer and Lungwitz[208] is based on the fact that in an ammoniacal solution, by means of barium oxalate, all the alkaline earths can be precipitated as oxalates, and sulfuric acid in similar circumstances can be thrown down as a barium salt and the iron and alumina as hydroxids. The reagents used to secure this precipitation are ammonia and barium oxalate.
For the determination of potash in a superphosphate the analytical process is conducted as follows: Ten grams of the superphosphate are mixed with half a liter of water and fifteen grams of barium oxalate dissolved in hydrochloric acid.
The mixture is boiled for twenty minutes and treated with some hydrogen peroxid to oxidize any ferrous iron that may be present. Afterwards the solution is made alkaline with ammonia. After cooling, it is made up to a given volume (half a liter) and filtered. An aliquot part of the filtrate is evaporated to dryness, ignited, extracted with hot water and, after the addition of a few drops of hydrochloric acid, the potassium is precipitated with platinic chlorid, and collected and weighed in the usual manner: Or the ignited residue may be dissolved directly in dilute hydrochloric acid and the rest of the process carried out as indicated.
In kainit the process is conducted as follows: Ten grams of the powdered sample are treated with a hydrochloric acid solution of the barium oxalate containing ten grams of the salt. The rest of the operation is conducted as described above. In the use of this method it is important that always enough of the barium oxalate solution be employed to fully saturate all the sulfuric acid which may be present.
261. Method of DeRoode for Kainit.—All the potash contained in kainit, according to de Roode, passes readily into aqueous solution.[209] On evaporating this aqueous solution to a pasty condition with enough platinic chlorid to unite with all the halogens present all the other bodies can be washed out of the potassium platinochlorid by ammonium [Pg 282] chlorid solution and the pure platinum salt thus obtained, which is washed and dried in the usual way. De Roode therefore asserts that it is quite useless to previously precipitate the solution of kainit with barium chlorid, ammonium oxalate, or carbonate. Before the addition of alcohol to the residue obtained by evaporation with platinic chlorid the sodium sulfate present renders the platinum salt sticky and difficult to wash, but the disturbing sodium compound can be readily removed by washing with ammonium chlorid solution.
The method of direct treatment has the advantage of avoiding the occlusion of potash in other precipitates and the danger of loss on ignition. The method as used by de Roode gives results about one-tenth per cent higher than are obtained by the official processes.
262. The Calcium Chlorid Method.—Huston has proposed the addition of calcium chlorid to the solution of a fertilizer in the determination of potash, in order to furnish sufficient calcium to form tricalcium phosphate with all the phosphoric acid present, and thereby permit of the use of platinum dishes in the lindo-gladding method.[210] In testing this process de Roode found that when sufficient calcium chlorid was added to combine with all the phosphoric acid present and then ammonia added in excess and a portion of the solution filtered, no test for phosphoric acid could be obtained; but, that if in addition to the calcium chlorid and ammonia, some ammonium oxalate or carbonate was added, a filtered portion of the solution gave a test for phosphoric acid.[211] This is accounted for by the fact that the calcium phosphate, which is precipitated by the ammonia, is changed by the ammonium oxalate or carbonate into calcium oxalate or carbonate and ammonium phosphate, so that the very object for which the calcium chlorid was added is defeated by the addition of the ammonium oxalate or carbonate. In order to make the use of calcium chlorid effective it is necessary to filter the liquid from the precipitate formed by the calcium chlorid and ammonia and then add the ammonium oxalate or carbonate to the filtrate. This necessitates two separate filtrations and makes the proposed method of Huston as long as the old process. [Pg 283]
263. Rapid Control Method for Potash Salts.—For rapid control work where great accuracy is not required Albert recommends that the finely ground substance be placed in a liter flask and about 400 cubic centimeters of water added and three cubic centimeters of hydrochloric acid.[212] After boiling, barium chlorid is added drop by drop as long as a precipitate is produced. After cooling, the flask is filled to the mark and shaken and its contents filtered through a dry filter. An aliquot portion of the filtrate is evaporated with platinum chlorid solution in a smooth porcelain dish almost to dryness and the mass treated with alcohol, filtered through a weighed filter, and well washed with alcohol. The filter is then dried in an air-bath to a constant weight. For the different kinds of potash materials on the market the following proportions are recommended:
Kainit or Carnallit.—Twenty grams in one liter: Fifty cubic centimeters of the filtrate are evaporated with forty of platinic chlorid solution. The weight of potassium platinochlorid obtained × 19.3 gives the per cent of K₂O.
Sulfate of Potash.—Fifteen grams in one liter: Twenty cubic centimeters of the solution are evaporated with fifteen of platinic chlorid. The weight of potassium platinochlorid obtained × 64.33 gives the per cent of K₂O.
Potassium Chlorid.—Ten grams in one liter: Twenty-five cubic centimeters are evaporated with fifteen of platinic chlorid solution. The weight of the precipitate obtained × 77.2 gives the per cent of K₂O.
264. Weighing the Precipitate as Metallic Platinum.—Hilgard calls attention to the difficulty of weighing the double chlorid of platinum and potash as such, although he acknowledges that in the gooch this weighing can be made with great accuracy.[213] He prefers to estimate the platinum in the metallic state and uses for this purpose a platinum crucible the inside of which, half way up from the bottom, is coated with a layer of platinum sponge, which is conveniently prepared by the decomposition of a few decigrams of the platinum double salt by inclining the crucible and rotating it during the progress of the reduction, using about a quarter of an hour in all. The platinum sponge produced in this way greatly favors [Pg 284] the decomposition of the double salt for analytical purposes. The decomposition of the salt takes place quickly and quietly and at conveniently low temperatures.
When the decomposition is ended the crucible is strongly heated so as to hold the platinum sponge, which is produced, together sufficiently to prevent its being removed in the subsequent washing of the crucible by decantation. By the ignition at a high temperature necessary to secure this, the greater part of the calcium chlorid is volatilized. After cooling, a few drops of concentrated hydrochloric acid are placed in the crucible and if the slightest yellow color be shown the acid is evaporated and the ignition repeated, with the addition of a little oxalic acid. In most cases the slight yellow color produced comes from a trace of iron and will therefore appear again after the second ignition. The crucible is subsequently washed by repeated decantations, finally with boiling water, and after drying is ignited and weighed.
The advantage of this process is that without further trouble the reduced metal is completely freed of any salts of the alkaline earths, etc., which have been carried down with it and also from any of the uncombined sodium chlorid which may not have been washed out by the alcohol. In fact, the results obtained in this way are nearly always lower than those obtained through the direct weighing of the double salt, and the wash water which is first poured off contains, as a rule, traces of the alkaline earths and almost without exception some sodium chlorid. Correction for the filter ash is unnecessary because the ash is completely dissolved by the treatment received. The platinum sponge which is collected in the crucible in this way is removed in case it does not adhere to the sides and the crucible is then ready for the next operation.
265. Sources of Error in the Platinum Method.—In the comparative work done in the determination of potash by the members of the Association of Official Agricultural Chemists there has been noted, from year to year, marked differences in the data obtained by different analysts. Such differences often are due to personal errors, or a failure to accurately follow the directions for manipulation. Sometimes, however, they are due to sources of error in the processes [Pg 285] employed. In the platinum method these sources of error have been long known to exist. Chief among these is the remarkable facility with which potash becomes incorporated with the precipitates of other bodies. The character and magnitude of some of these errors have lately been studied by Robinson.[214]
Many precipitates occlude potash and hold it so firmly that it cannot be washed out with hot water although the potash compounds present in the precipitate are perfectly soluble. It appears to be a kind of molecular adhesion. Barium sulfate has this property of attaching potash molecules in a high degree, and ferric and aluminic compounds only to a slightly less extent. To reduce the losses, consequent on the conditions just mentioned, to a minimum, the sulfuric acid and earthy bases should be very slowly precipitated, with violent agitation, at a boiling temperature.
Another source of loss in the platinum method arises from the use of a solution of ammonium chlorid for washing the potassium platinochlorid precipitate. There is danger here, not only of the solution of the impurities present in the precipitate, but also of a double decomposition by means of which some ammonium may be substituted for the potassium in the washed product. In the official method, moreover, there is danger of securing a final precipitate which may contain traces of calcium and magnesium sulfates when these bodies are abundantly present in the sample taken for analysis. The careful analyst must guard against these sources of error, but it is probably true that he will never secure a practically chemically pure precipitate of potassium platinochlorid when working on the mixed fertilizers found in commerce.
266. Effect of Concentration on the Accuracy of Potash Analysis.—Winton has also studied the sources of error in the determination of potash as platinochlorid, especially with reference to the effect of the concentration of the solution at the time of precipitation.[215]
He finds that the method of precipitating in concentrated solutions and drying the potassium platinochlorid at 130°, depends for its accuracy upon the mutual compensation of three errors; viz., (1) to the [Pg 286] solubility of the potassium salt in eighty per cent alcohol, (2) to the presence of water in the crystals which is not driven off at 130°, and (3) the use of a factor based on the wrong atomic weight of platinum.
He finds, further, that the error due to the presence of water occluded in the crystals can be reduced to a minimum, and the process of drying greatly simplified, by adding the solution of platinum chlorid to the potash solution in a dilute condition, not exceeding one per cent in strength. The potassium platinochlorid thus produced can be very effectively dried at 100°. The error due to the solubility of the salt in eighty per cent alcohol can also be greatly reduced by using ninety-five per cent alcohol. The error due to the wrong factor, based on the old atomic weight of platinum, viz., 0.3056, can be corrected by using the factor based on the recently determined atomic weight of platinum, viz., 195, which is 0.30688.
267. Differences in Crystalline Form.—Winton has also observed a distinct difference in the crystals of potassium platinochlorid when obtained from concentrated and dilute solutions.[216] When platinic chlorid is added to a concentrated solution of potassium chlorid, a large part of the salt which is formed is precipitated in a pulverulent state, the remainder being deposited on evaporation. After treating with alcohol, filtering, and drying, the double salt is found in the state of a fine powder which, when examined under the microscope, is found to consist largely of radiating crystals. The characteristic form is one having six arms formed by the intersection, at right angles, of three bars. Numerous globular cavities in the crystals are observed in which mother liquid is enclosed. For this reason the salt is not easily dried at 100°, but when so dried loses additional moisture at 130°, and still more at 160°. The total additional loss, after drying at 100°, from this cause may amount to as much as six-tenths per cent of potassium chlorid.
When, however, the solution of the potassium salt is so dilute that no precipitate at all is formed on the addition of platinic chlorid, the double salt is all deposited, as well as formed slowly, during the evaporation and occurs exclusively as octahedra. These octahedra are [Pg 287] comparatively free of cavities, and give up practically all their moisture when dried at 100°. A method of procedure therefore for potash determination, based on the above principle of the addition of the reagent to dilute solutions, and drying the double salt produced upon evaporation, after washing with ninety-five per cent alcohol at 100°, and using the factor 0.30688 for potassium chlorid and 0.1939 for potassium oxid, gives good results and is regarded as better than any of the methods which prescribe the addition of platinic chlorid to highly concentrated potash solutions.
268. Factors for Potash Estimation.—The factor now in use by the official chemists to convert potassium platinochlorid into potash (K₂O) is 0.19308, and for potassium chlorid 0.3056.
Wolfbauer gives the differences which may arise by computing the potash from its platino-double chlorid by the different values assigned to the atomic weight of platinum.[217]
The common factor used to obtain potassium chlorid from potassium platinochlorid is based on the atomic weight 197.18 and is derived from the formula:
| 2(39.13 + 35.46) | = | 149.18 | = 0.30557. |
| 2 × 39.13 + 197.18 + 6 × 35.46 | 488.20 |
The variations arising from taking other assigned values for the atomic weight of platinum are shown in the following table:
| Factor for potassium chlorid from |
Relation to factor 0.30557 in per cent |
|||||
|---|---|---|---|---|---|---|
| Atomic weight of Platinum. |
Determined or calculated by |
Potassium platinochlorid. |
Platinum. | Potassium platinochlorid. |
Platinum. | |
| 197.18 | Berzelius | 0.30557 | 0.75658 | 100.00 | 100.00 | |
| 197.88 | Andrews | 0.30517 | 0.75390 | 99.86 | 99.65 | |
| 195.06 | Haberstadt | 0.30690 | 0.76468 | 100.44 | 101.07 | |
| 194.87 | Seubert and | 0.30700 | 0.76555 | 100.47 | 101.20 | |
| Clark | ||||||
The factor 0.3056 is regarded as the best for the computation from potassium platinochlorid and 0.7566 from platinum. It is also suggested that it is better to make the computation from the reduced platinum than from the double salt.
269. Recovery of the Platinum Waste and Preparation of the Platinic Chlorid Solution.—(1) By Reduction in Alkaline Alcohol.—All filtrates containing platinic chlorid, all precipitates of potassium platinochlorid and all residues of metallic [Pg 288] platinum should be carefully preserved and the platinum recovered therefrom by the following process: The platinum residues are placed in a large porcelain dish. Since these residues contain a large amount of alcohol they should be diluted with about one-third their volume of water, and when boiling treated with some sodium carbonate. The solid potassium platinochlorids should not be added until the liquid is boiling, and then only little by little. The heating on the water-bath is continued until the liquid floating over the platinum sponge is quite clear and only slightly yellow. The liquid is then poured off and the reduced platinum purified by boiling with hydrochloric acid and water. It is then dried and ignited to destroy any organic matter which may be present. It is advisable to boil the finely divided platinum once with strong nitric acid, and after this is poured off the solution of the platinum is effected in a large porcelain dish over a water-bath by adding about four times its weight of hydrochloric acid, warming, and adding nitric acid, little by little. After the platinum is in solution the evaporation is continued until a drop of the liquid, removed by a glass rod, quickly solidifies. The crystalline mass which is formed on cooling is taken up with water and filtered, and then a sufficient amount of water added so that each ten cubic centimeters will contain one gram of platinum. The specific gravity of this solution is 1.18 at ordinary temperatures. Special care must be taken that the solution contains neither platinous chlorid nor nitrogen compounds. If the first named compound be present it should be converted into platinic chlorid by treatment with fuming hydrochloric acid and a little nitric acid. The last mentioned compound may be removed by evaporating successively with hydrochloric acid and water. If the platinic chlorid be made from waste platinum, the danger of contamination with iridium must be considered. In such a case the platinum should be separated as ammonium platinochlorid, which can afterwards be reduced as above indicated. A convenient test of the purity of platinic chlorid solution is accomplished by the precipitation of a known weight of chemically pure potassium salt.
(2) By Reduction in Nascent Hydrogen.—The platinum residues, filtrates containing platinum, etc., are collected in a large flask and [Pg 289] evaporated in a large dish on a water-bath, and reduced by means of zinc and hydrochloric acid to metallic platinum, the mass being warmed until all the zinc has been dissolved. The supernatant liquid standing over the spongy platinum is decanted and the spongy mass boiled twice with distilled water. The spongy platinum is then brought on a filter and washed till the filtrate shows no acid reaction. The filter and platinum sponge are next incinerated in a platinum dish and the residue weighed. The weighed mass of pure platinum is dissolved in hydrochloric acid, with the addition of as little nitric acid as possible, and, after cooling, filtered. The filtrate is afterwards evaporated in a porcelain dish on a water-bath to a sirupy consistence, taken up with water and filtered. To this filtrate enough water is now added to make the solution correspond to one gram of metallic platinum in ten cubic centimeters.
270. General Principles.—By reason of the great cost of platinum chlorid analysts have sought for a reagent of a cheaper nature and yet capable of forming an insoluble compound with potash. Phosphomolybdic and perchloric acids are the reagents which have given the most promising results.[218] The principle of the method with the latter salt is based on the insolubility of potassium perchlorate in strong alcohol containing a little perchloric acid and the comparative easy solubility of the other bases usually associated with potassium in water. The French chemists have stated that magnesia, when present in considerable quantities, interferes with the accuracy of the results. Since in soil analysis considerable quantities of magnesia are often found, this base, according to the French chemists, should previously be removed when present in any considerable quantity, by the process described in the first volume. Kreider, however, as will be seen further on, working in the presence of magnesia, did not notice any disturbing effects caused thereby. The method is applicable to the common potash salts of the trade and with certain precautions to mixed salts. As will be mentioned later on, sulfuric acid should be previously removed and this is likely to introduce an error on account of the tendency of barium sulfate to [Pg 290] entangle particles of potash among its molecules and thus remove them from solution. The barium sulfate should be precipitated slowly and in a strongly acid (nitric or hydrochloric) solution. The loss, which is inevitable, is thus reduced to a minimum and does not seriously affect the value of the numbers found. It is important to have an abundant supply of pure perchloric acid, and as this is not readily obtainable in the market the best methods of preparing it are given below. The method, while it has not been worked out extensively, is one of merit, and seemingly is worthy of fair trial by analysts. The process is by no means a new one, but it will not be necessary to describe here its development any further than to refer to the methods proposed by Serullas,[219] Schlösing,[220] Kraut,[221] and Bertrand,[222] The method was fully developed by a committee appointed by the French agricultural chemists in 1887.[223]
Wense has also described an improved method of estimating potash as perchlorate after the removal of sulfuric acid and also a process of preparing perchloric acid by distilling potassium perchlorate with sulfuric acid in a vacuum.[224] He was also the first who proposed the plan of rendering potassium perchlorate insoluble in alcohol by dissolving a little perchloric acid therein.[225] The best approved methods now known of preparing the perchloric acid and conducting the analysis will be described in the following paragraphs.
271. Caspari’s Method for Preparing Perchloric Acid.—A hessian crucible about fifteen centimeters high is filled with moderately well compressed pure potassium chlorate and gradually heated in a suitable furnace until the contents become fluid.[226] The heat must then be carefully regulated to avoid loss by foaming due to the evolution of oxygen. The heat is continued until oxygen is no longer given off and the surface of the liquid becomes encrusted, which will take place in from one and a half to two hours.
After cooling, the contents of the crucible are pulverized and heated, with vigorous stirring, to boiling, with one and a half times their weight of water. By this process the potassium chlorid which has formed during the first reaction is dissolved and is thus removed. The residual salt is washed with additional quantities of cold water and [Pg 291] finally dried. To remove the potassium salt from the crude potassium perchlorate obtained as above, recourse is had to hydrofluosilicic acid. The reaction is represented by the following formula: 2KClO₄ + H₂SiF₆ = K₂SiF₆ + HClO₄. In order to effect this decomposition the potassium perchlorate is dissolved in seven times its weight of hot water and an excess of hydrofluosilicic added to the boiling solution. The boiling is continued for about an hour until particles of potassium perchlorate can no longer be detected with addition of water to compensate for evaporation.
On cooling the gelatinous potassium silicofluorid is deposited and the perchloric acid separated therefrom as completely as possible by decantation. The residue is again boiled with water and a little hydrofluosilicic acid and the clear liquor thus obtained added to the first lot. Finally, any residual perchloric acid may be removed on an asbestos felt under pressure. The clear liquid thus obtained is evaporated on a steam-bath to the greatest possible degree of concentration and allowed to stand in a cool place for twenty-four hours, whereby is effected the separation of any remaining potassium silicofluorid or potassium perchlorate. The residual liquid when filtered through an asbestos felt should give a perfectly clear filtrate. In order to throw out the last traces of hydrofluosilicic acid and any sulfuric acid present an equal volume of water is added, and while cold small quantities of barium chlorid are successively added until the barium salt is present in a very slight excess. The clear supernatant liquid is poured off after a few hours and evaporated until the hydrochloric acid is all expelled and white fumes of perchloric acid are noticed. Any potassium perchlorate still remaining will now be separated and, in the cold, sodium perchlorate will also be separated in crystals. The clear residue is again diluted with an equal volume of water and any barium salts present carefully removed with sulfuric acid. The mass is allowed to stand for one or two days, and is then filtered through paper and is ready for use. The purity of the acid obtained depends chiefly on the purity of the hydrofluosilicic acid at first used. Hence to get good results this acid must be free from foreign bodies. If an absolutely pure product be desired the acid above obtained must be distilled in a vacuum. [Pg 292]
272. Method of Kreider.—Kreider has worked out a simpler method of preparing perchloric acid which will make it easy for every analyst to make and keep a supply of this admirable yet unappreciated reagent. This method is conducted as follows:[227]
A convenient quantity of sodium chlorate, from 100 to 300 grains, is melted in a glass retort or round-bottomed flask and gradually raised to a temperature at which oxygen is freely, but not too rapidly evolved, and kept at this temperature till the fused mass thickens throughout, indicating the complete conversion of the chlorate to the chlorid and perchlorate, which requires from one and one-half to two hours: or the retort may be connected with a gasometer and the end of the reaction determined by the volume of oxygen expelled, according to the equation
2NAClO₃ = NACl + NAClO₄ + O₂.
The product thus obtained is washed from the retort to a capacious evaporating dish where it is treated with sufficient hydrochloric acid to effect the complete reduction of the residual chlorate, which, if the ignition has been carefully conducted with well distributed heat, will be present in but small amount. It is then evaporated to dryness on the steam-bath, or more quickly over a direct flame, and with but little attention until a point near to dryness has been reached, when stirring will be found of great advantage in facilitating the volatilization of the remaining liquid and in breaking up the mass of salt. Otherwise the perchlorate seems to solidify with a certain amount of water and its removal from the dish, without moistening and reheating, is impossible.
After triturating the residue, easily accomplished in a porcelain mortar, an excess of the strongest hydrochloric acid is added to the dry salt, preferably in a tall beaker where there is less surface for the escape of hydrochloric acid and from which the acid can be decanted without disturbing the precipitated chlorid. If the salt has been reduced to a very fine powder, by stirring energetically for a minute, the hydrochloric acid will set free the perchloric acid and precipitate the sodium as chlorid, which in a few minutes settles, leaving a clear solution of the perchloric acid with the excess of hydrochloric acid. [Pg 293] The clear supernatant liquid is then decanted upon a gooch, through which it may be rapidly drawn with the aid of suction, and the residue retreated with the strongest hydrochloric acid, settled, and again decanted, the salt being finally brought upon the filter where it is washed with a little strong hydrochloric acid. A large platinum cone will be found more convenient than the crucible, because of its greater capacity and filtering surface. When the filter will not hold all the sodium chlorid, the latter, after washing, may be removed by water or by mechanical means, with precautions not to disturb the felt, which is then ready for the remainder. Of course, if water is used, the felt had better be washed with a little strong hydrochloric acid before receiving another portion of the salt. This residue will be found to contain only an inconsiderable amount of perchlorate, when tested by first heating to expel the free acid and then treating the dry and powdered residue with ninety-seven per cent alcohol, which dissolves the perchlorate of sodium but has little soluble effect on the chlorid.
The filtrate, containing the perchloric acid with the excess of hydrochloric acid and the small per cent of sodium chlorid which is soluble in the latter, is then evaporated over the steam-bath till all hydrochloric acid is expelled and the heavy white fumes of perchloric acid appear, when it is ready for use in potassium determinations. Evidently the acid will not be chemically pure because the sodium chlorid is not absolutely insoluble in hydrochloric acid; but a portion tested with silver nitrate will prove that the sodium, together with any other bases which may have gone through the filter, has been completely converted into perchlorate, and unless the original chlorate contained some potassium or on evaporation the acid was exposed to the fumes of ammonia, the residue of the evaporation of a portion is easily and completely soluble in ninety-seven per cent alcohol and its presence is therefore unobjectionable. One cubic centimeter of the acid thus obtained gives on evaporation a residue of only 0.036 gram, which is completely soluble in ninety-seven per cent alcohol.
Caspari’s acid under similar treatment gave a residue in one case of [Pg 294] 0.024 gram and in another 0.047 gram. If, however, a portion of pure acid be required, it may be obtained by distilling this product under diminished pressure and, as Caspari has shown, without great loss providing the heat is regulated according to the fumes in the distilling flask.
Some modification of the above treatment will be found necessary in case the sodium chlorate contains any potassium as an impurity, or if the latter has been introduced from the vessel in which the fusion was made. In these circumstances the hydrochloric acid would not suffice for the removal of potassium, since a trace might also go over with the sodium and thus on evaporation a residue insoluble in ninety-seven per cent alcohol be obtained. To avoid this difficulty, the mixture of sodium perchlorate and chlorid, after treating with hydrochloric acid for the reduction of the residual chlorate, being reduced to a fine powder, is well digested with ninety-seven per cent alcohol, which dissolves the sodium perchlorate but leaves the chlorid, as well as any potassium salt insoluble. By giving the alcohol time to become saturated, which was facilitated by stirring, it was found on filtering and evaporating that an average of about two-tenths of a gram of sodium perchlorate are obtained for every cubic centimeter of alcohol and that the product thus obtained is comparatively free of chlorids, until the perchlorate is nearly all removed, when more of the chlorid seems to dissolve. This treatment with alcohol is continued until on evaporation of a small portion of the latest filtrate, only a small residue is found. The alcoholic solution of the perchlorate is then distilled from a large flask until the perchlorate begins to crystallize, when the heat is removed and the contents quickly emptied into an evaporating dish, the same liquid being used to wash out the remaining portions of the salt. When the distillation is terminated at the point indicated, the distillate will contain most of the alcohol employed, but in a somewhat stronger solution, so that it requires only diluting to ninety-seven per cent to fit it for use in future preparations. The salt is then evaporated to dryness on the steam-bath and subsequently treated with strong hydrochloric acid for the separation of the perchloric acid. [Pg 295]
One cubic centimeter of the acid prepared in this way, on evaporation gave a residue in one case of 0.0369 gram, and in another 0.0307 gram, completely soluble in ninety-seven per cent alcohol, which was then ignited and the chlorin determined by silver from which the equivalent of perchloric acid in the form of salts was calculated as 0.0305 gram. By neutralizing the acid with sodium carbonate, evaporating, igniting in an atmosphere of carbon dioxid till decomposition was complete, collecting the oxygen over caustic potash, allowing it to act on hydriodic acid by intervention of nitric oxid, according to a process soon to be published, titrating the iodin liberated, with standard arsenic and calculating the equivalent of perchloric acid, after subtracting the amount of acid found in the form of salts, the amount of free acid per cubic centimeter proved to be 0.9831 gram.
The whole process, even when the separation with alcohol is necessary, can not well require more than two days and during the greater part of that time the work proceeds without attention.
273. Keeping Properties of Perchloric Acid.—By most authorities it is asserted that perchloric acid is a very unstable body and is liable to decompose with explosive violence even when kept in the dark. It is probable that this tendency to spontaneous decomposition has been exaggerated. It is not even mentioned in Gmelin’s Handbook.[228]
The most concentrated aqueous acid has a specific gravity of 1.65, is colorless, fumes slightly when exposed to the air, and boils at 200°. It has no odor, possesses an oily consistence and has a strong and agreeably acid taste. It reddens litmus without bleaching it and is slowly volatilized at 138° without decomposition. It is unaffected by exposure to the light, even the sun’s rays. It is not decomposed by hydrosulfuric, sulfurous, or hydrochloric acids, nor by alcohol. Paper saturated with the strong acid does not take fire spontaneously, but it deflagrates with red-hot charcoal.
The acid prepared by the method of Kreider has approximately the composition of the di-hydrate, HClO₄·2H₂O.[229] Unless well evaporated, however, it is a little more dilute than is shown by the above formula. The di-hydrate is quite stable and the more [Pg 296] dilute acid can be kept for an indefinite time. Kreider has kept the acid for six months and noticed no change whatever in its composition. Acid containing one gram of perchloric acid in a cubic centimeter has been kept three months with perfect safety. There is no reason why the strong aqueous acid should not be made a regular article of commerce by dealers in chemical supplies, under proper restrictions for storage and transportation.
The strong acid made in this laboratory by the Kreider method has not given the least indication of easy or spontaneous decomposition.
274. The Analytical Process.—The perchlorate process cannot be applied in the presence of sulfuric acid or dissolved sulfates. This acid, when present, is to be removed by the usual methods before applying the perchloric acid. Phosphoric acid may be present, but in this case a considerable excess of the reagent must be used. The process, as originally proposed by Caspari and carried out by Kreider, is as follows:[230]
The substance, free from sulfuric acid, is evaporated for the expulsion of free hydrochloric acid, the residue stirred with twenty cubic centimeters of hot water and then treated with perchloric acid, in quantity not less than one and one-half times that required by the bases present, when it is evaporated, with frequent stirring, to a thick, sirup-like consistency, again dissolved in hot water and evaporated, with continued stirring, till all hydrochloric acid has been expelled and the fumes of perchloric acid appear. Further loss of perchloric acid is to be compensated for by addition of more. The cold mass is then well stirred with about twenty cubic centimeters of wash alcohol—ninety-seven per cent alcohol containing two-tenths per cent by weight of pure perchloric acid, with precautions against reducing the potassium perchlorate crystals to too fine a powder. After settling, the alcohol is decanted on the asbestos filter and the residue similarly treated with about the same amount of wash alcohol, settled, and again decanted. The residual salt is then deprived of alcohol by gently heating, dissolved in ten cubic centimeters of hot water and a little perchloric acid, when it is evaporated once more, [Pg 297] with stirring, until fumes of perchloric acid rise. It is then washed with one cubic centimeter of wash alcohol, transferred to the asbestos, preferably by a policeman to avoid excessive use of alcohol, and covered finally with pure alcohol; the whole wash process requiring from about fifty to seventy cubic centimeters of alcohol. It is then dried at about 130° and weighed.
The substitution of a gooch for the truncated pipette employed by Caspari will be found advantageous; and asbestos capable of forming a close, compact felt should be selected, inasmuch as the perchlorate is in part unavoidably reduced, during the necessary stirring, to so fine a condition that it tends to run through the filter when under pressure. A special felt of an excellent quality of asbestos was prepared for the determinations given below and seemed to hold the finer particles of the perchlorate very satisfactorily.
A number of determinations made of potassium, unmixed with other bases or non-volatile acids, is recorded in the following table:
| Potassium chlorid taken. |
Volume of filtrate. |
Potassium perchlorate found. |
Error on potassium perchlorate. |
Error on potassium chlorid. |
Error on potash. |
|---|---|---|---|---|---|
| Grams. | Cubic centimeters. |
Grams. | Grams. | Grams. | Grams. |
| 0.1000 | 54 | 0.1851 | 0.0008— | 0.0004— | 0.0003— |
| 0.1000 | 58 | 0.1854 | 0.0005— | 0.0002— | 0.0002— |
| 0.1000 | 51 | 0.1859 | 0.0000 | 0.0000 | 0.0000 |
| 0.1000 | 50 | 0.1854 | 0.0005— | 0.0002— | 0.0002— |
| 0.1000 | 48 | 0.1859 | 0.0000 | 0.0000 | 0.0000 |
| 0.1000 | 52 | 0.1854 | 0.0005— | 0.0002— | 0.0002— |
Considerable difficulty, however, was experienced in obtaining satisfactory determinations of potassium associated with sulfuric and phosphoric acids. As Caspari has pointed out, the sulfuric acid must be removed by precipitation as barium sulfate before the treatment with perchloric acid is attempted, and unless the precipitation is made in a strongly acid solution, some potassium is carried down with the barium. Phosphoric acid need not be previously removed, but to secure a nearly complete separation of this acid from the potassium, a considerable excess of perchloric acid should be left upon the potassium perchlorate before it is treated with the alcohol. When these conditions are [Pg 298] carefully complied with, fairly good results may justly be expected. Below is given a number of the results obtained:
| Compounds taken. |
(A) | (B) | (C) | (D) | (E) |
|---|---|---|---|---|---|
| Grams | Cubic centimeters. |
Grams. | Grams. | Grams. | Grams. |
| Potassium chlorid = 0.10 | |||||
| Calcium carbonate = 0.13 | 50 | 0.1887 | 0.0027+ | 0.0014+ | 0.0005+ [231] |
| Magnesium sulfate = 0.13 | 82 | 0.1875 | 0.0016+ | 0.0008+ | 0.0005+ [232] |
| Ferric chlorid = 0.05 | 80 | 0.1861 | 0.0002+ | 0.0001+ | 0.0001+ [233] |
| Magnesium sulfate = 0.05 | 80 | 0.1843 | 0.0016- | 0.0008- | 0.0005- [234] |
| Manganese dioxid = 0.05 | 92 | 0.1839 | 0.0020- | 0.0010- | 0.0006- [235] |
| Sodium phosphate = 0.40 | 60 | 0.1854 | 0.0005- | 0.0002- | 0.0002- [236] |
In the last three experiments of the above table the amount of perchloric acid was about three times that required to unite with the bases present, and the phosphoric acid subsequently found with the potassium was hardly enough to appreciably affect the weight, although its absolute removal was found impossible.
That the magnesia does not produce any disturbing effect, as is supposed by the French chemists, Kreider has proved by the following test: One hundred and fifty milligrams of magnesium carbonate were treated with perchloric acid, evaporated till fumes of perchloric acid appeared, and cooled, when the magnesium perchlorate crystallized: But on treating it with about fifteen cubic centimeters of ninety-seven per cent alcohol containing two-tenths per cent of perchloric acid a perfectly clear solution was obtained. If, therefore, a sufficient excess of acid be used, no interference will be caused by the presence of magnesium.
While it is true, therefore, that the potassium perchlorate obtained may be contaminated with a trace of phosphoric acid, if the latter be present in large quantity, no fear of contamination with magnesia need be entertained if a sufficient quantity of the perchloric acid be used.
275. Removal of the Sulfuric Acid.—The practical objection to the removal of the sulfuric acid in the form of barium sulfate rests on the fact of the mechanical entanglement of some of the potash in the barium salt. Unless special precautions are taken, therefore, a [Pg 299] considerable amount of the potash will be found with the barium sulfate.
Caspari has succeeded in reducing this amount to a minimum by the following procedure:[237] The solution of barium chlorid is prepared by dissolving 127 grams of crystallized barium chlorid in water, adding 125 cubic centimeters of thirty-five per cent hydrochloric acid, and bringing the total volume up to one liter with water.
Five grams of the substance from which the sulfuric acid is to be removed are boiled with 150 cubic centimeters of water and twenty of strong hydrochloric acid. While the solution is still in ebullition it is treated, drop by drop with constant stirring, with the barium chlorid solution above mentioned, until a slight excess is added. This excess does not cause any inconvenience subsequently. After the precipitation is complete the boiling is continued for a few minutes, the mixture cooled and made up to a quarter of a liter with water. No account is taken of the volume of the barium sulfate formed since, even with the precautions mentioned, a little potassium is thrown down and the volume of the barium sulfate tends to correct this error. With a solution from which the sulfuric acid had been removed as above indicated, Caspari found a loss of only one milligram of potassium perchlorate in a precipitate weighing over 800 milligrams.
276. Applicability of the Process.—Experience has shown that sulfuric acid is the only substance which need be removed from ordinary fertilizers preparatory to the estimation of the potash by means of perchloric acid. The fact that this process can be used in the presence of phosphoric acid is a matter of great importance in the estimation of potash in fertilizers, inasmuch as these fertilizers nearly always contain that acid. The fact that the French chemists noticed that magnesia was a disturbing element in the process, as has been indicated in volume first, probably arose from its presence as sulfate. Neither Caspari nor Kreider has noticed any disturbance in the results which can be traced to the presence of magnesia as a base.
If ammonia be present, however, there is a tendency to the production of ammonium perchlorate which is somewhat insoluble in the alcohol wash used. Solutions therefore containing ammonia before treating [Pg 300] by the perchlorate method for potash should be rendered alkaline by soda-lye and boiled. With the precautions above mentioned, the method promises to prove of great value in agricultural analysis, effecting both a saving of time and expense in potash determinations.
277. Accuracy of the Process.—The perchlorate was tried in conjunction with the platinum method on the two samples of potash fertilizer prepared and distributed by the official reporter on potash for 1893.[238] One of the samples was of a fertilizer which had been compounded for the Florida trade and contained bone, dried blood, and potash, mostly in the form of sulfate. The other sample consisted of mixed potash salts, sulfate, chlorid, double salt, kainit, and about five per cent of the triple sulfate of calcium, potassium, and magnesium.
The results obtained by Wagner and Caspari on the two samples follow:
| Sample No. 1. Per cent potash. |
Sample No. 2. Per cent potash. |
|
|---|---|---|
| By the platinum method | 13.25 | 37.98 |
| By the perchlorate method | 13.09 | 37.82 |
The perchlorate method on the whole appears to be quite as accurate as the platinum process, requires less manipulation and can be completed in a shorter time and at less expense for reagents.
[185] Connecticut Agricultural Experiment Station, Bulletin No. 97, p. 7.
[186] Annual Report Connecticut Station, 1892, p. 32.
[187] Colorado Agricultural Experiment Station, Bulletin No. 10.
[188] Connecticut Agricultural Experiment Station, Bulletin No. 103, p. 9.
[189] Annual Report, Massachusetts Agricultural Experiment Station, 1888, p. 202.
[190] Vid. op. cit. 2, 1890, p. 110.
[191] Traité de la Fabrication de Sucre, Horsin-Déon, p. 511.
[192] Chemical Division, U. S. Department of Agriculture, Bulletin No. 37, p. 350.
[193] Journal of the American Chemical Society, Vol. 17, p. 86.
[194] Volume First, pp. 19, et seq.
[195] Precht: Die Stassfurter Kalisalze.
[196] Maercker: Die Kalidüngung, S. 1.
[197] Vid. op. cit. supra, p. 3.
[Pg 301]
[198] Vid. op. cit. 12, p. 5.
[199] Vid. op. cit. 12, p. 7.
[200] Volume First, pp. 378, et seq.
[201] Chemical News, Vol. 44, pp. 77, 86, 97, and 129.
[202] Chemical Division, U. S. Department of Agriculture, Bulletin No. 7, p. 38.
[203] Vid. supra, Bulletin No. 43, p. 349.
[204] Die Agricultur-Chemische Versuchs-Station, Halle a/S., S. 76.
[205] Methoden van onderzock aan de Rijkslandbouw-proefstations, 1893, p. 7.
[206] From the Official Swedish Methods. Translated for the Author by F. W. Woll.
[207] Chemical Division, U. S. Department of Agriculture, Bulletin No. 35, p. 63.
[208] Chemiker Zeitung, Band 18, S. 1320.
[209] Journal of the American Chemical Society, Vol. 17, p. 85.
[210] Chemical Division, U. S. Department of Agriculture, Bulletin No. 43, p. 26.
[211] Vid. op. cit. 25, Vol. 17, p. 46.
[212] Zeitschrift für angewandte Chemie, 1891, S. 281.
[213] Zeitschrift für analytische Chemie, Band 32, S. 184.
[214] Vid. op. cit. 25, Vol. 16, p. 364.
[215] Vid. op. cit. 25, Vol. 17, p. 463.
[216] Vid. op. et. loc. cit. supra.
[217] Chemiker Zeitung, 1890, S. 1246.
[218] Volume First, pp. 369, 375.
[219] Annales de Chimie et de Physique {2}, Tome 46, p. 294.
[220] Comptes rendus, Tome 73, p. 1296.
[221] Zeitschrift für analytische Chemie, Band 14, S. 152.
[222] Chemical News, Vol. 44, p. 316.
[223] Rapport adressé par la Comité des Stations Agronomiques, 1887, p. 10.
[224] Zeitschrift für angewandte Chemie, 1891, S. 691.
[225] Vid. op. cit. supra, 1892, S. 233.
[226] Vid. op. cit. 40, 1893, S. 68.
[227] American Journal of Science, June, 1895, from advance proofs sent by author.
[228] Watt’s Translation, Vol. 2, pp. 317-318.
[229] Manuscript communication from Mr. Kreider.
[230] Vid. op. et loc. cit. 43.
[231] The residue showed phosphoric acid plainly when tested.
[232] The residue showed phosphoric acid plainly when tested.
[233] Only traces of phosphoric acid found in the residue.
[234] Only traces of phosphoric acid found in the residue.
[235] Only traces of phosphoric acid found in the residue.
[236] Only traces of phosphoric acid found in the residue.
[237] Zeitschrift für angewandte Chemie, 1893, S. 73.
[238] Chemical Division, U. S. Department of Agriculture, Bulletin No. 38, p. 57.
[Pg 302]
278. Classification.—Nitrogen, phosphoric acid, and potash are the most important of the plant foods both from a commercial and physiological point of view. They are the chief constituents of the most important fertilizers and manures, but are by no means the sole essential elements of plant nutrition. Lime, magnesia, soda, sulfur, chlorin, and many other elements are found constantly in plants and must be regarded as normal constituents thereof. It is the purpose here, however, to speak only of those substances which are used as fertilizers and which constantly or occasionally are subjected to chemical examination for determining their commercial or agronomic value. These bodies may be conveniently divided into two classes; viz., mineral and organic. Among those of mineral nature may be mentioned lime, gypsum, marls, wood-ashes, common salt, and ferrous sulfate; among those of organic nature may be included guano, hen manure, stall manure, composts, and muck.
279. Forms of Lime.—By the term lime is meant the product obtained by subjecting limestone or other lime carbonates to the action of heat until the carbon dioxid contained therein is expelled. The resulting lime, CaO, when exposed for some time to the air, or at once on the addition of water, is converted into the hydrate CaO₂H₂, known as slaked lime. On longer exposure to the air, the hydrate gradually absorbs carbon dioxid and becomes converted into carbonate. In whatever form, therefore, lime is applied to the soil, it is found in the end, as carbonate. A distinction should also be made between lime obtained from mineral substances and that got from organic products such as shells. Strictly speaking this is not a definite matter, inasmuch as limestones are sometimes but little more than aggregations of fossil shells. Practically, however, the distinction is made, and some farmers prefer shell lime to that of any other kind. Gas lime, that is lime [Pg 303] which has been used for the purification of illuminating gas made from coal, is hardly to be considered in this connection, since it may contain very little even of the hydrate. In this case the lime has been converted largely into carbonate and sulfid.
280. Application of Lime.—For many reasons it is important that the lime be transported to the field before it has had time to be converted into hydrate. The transportation costs less in this state and it can be handled with far less inconvenience than when slaked. The lime should be placed in small piles and left thus, best covered with a little earth, until thoroughly slaked. It is then spread evenly over the surface. The quantity used per acre depends largely on the nature of the soil. Stiff clays and sour marsh lands require a larger dressing than loams or well aerated soils. From three to six thousand pounds per acre are the quantities usually employed. When the lime is once thoroughly incorporated in the soil it is rapidly converted into carbonate, but while in the caustic state it may act vigorously in promoting the decay of organic matter and may prove injurious in promoting the decomposition of ammonium salts with attendant loss of nitrogen.
281. Action of Lime.—The benefits arising from the application of lime to agricultural lands, although in many cases great, do not arise from any distinct fertilizing action of its own. Plants need lime for growth and need plenty of it, but as a rule any soil which is good enough to grow crops will contain enough lime to furnish that constituent of the crops for many years. Its action is both mechanical and chemical. By virtue of the latter property it renders available for plant food bodies already existing in the soil but existing in such shape as to be unavailable for plants. The supply of plant food available for the crops of one year is increased but this increase is at the expense of the following years. Lime is a stimulant. There is an European proverb that “lime enriches the father but beggars the son.” Nevertheless, a limestone country is usually a fertile one and soils containing plenty of lime naturally, are nearly always rich soils. It is said that the trees and plants which farmers pick out as indicative of rich land are nearly always those which prefer lime soils. [Pg 304]
The mechanical action of lime on soils tends to lighten heavy clays and loams and to render firmer and more consistent the light and shifting sandy soils. When a lump of clay is stirred up in a bucket of rain water the water becomes muddy and remains that way for many days. If, however, to the bucket of muddy water a little lime water be added the suspended particles of clay begin to flocculate and soon the water is clear and the clay falls to the bottom, nor does it again make the water muddy for a long time when stirred up with it. The flocky character of the precipitate is tenaciously retained and it is necessary to knead the clay for some time to induce it to reassume its original heavy character. An action like this takes place when lime is added to heavy soils so that the soil becomes more porous and assumes a better tilth on plowing. With sandy soils an altogether different action takes place. In making mortar, as is well known, sand is stirred in with milk of lime and after being exposed to air for a while the mixture becomes hard and firm, the firmness increasing with age. This is due to the fact that when the mortar dries the lime begins to absorb carbon dioxid from the air and is converted into grains of carbonate which adhere strongly to neighboring sand grains and to each other so that the whole soon gets to be a solid mass. Something like this takes place in the soil and the sand grains are to some extent bound together. The increased firmness of the soil thus gained is often of considerable advantage.
Besides these actions, which are more or less mechanical, lime exerts a chemical action on many soil constituents. Feldspar and other common rocks contain potash, and this potash is in such a form as to be inaccessible to plants. These rocks exist in the shape of small particles in many soils and on them lime exerts a decomposing action, setting the potash free. Lime also hastens the decomposition of the nitrogenous organic matter and at the same time renders the soil more retentive of the products formed. The conversion of ammonia, resulting from the decomposition of such organic matter into nitrites and nitrates, is not easily accomplished without a proper amount of calcium carbonate. The microorganisms producing this change, which is known as nitrification, apparently require its presence for neutralizing the acid [Pg 305] formed. In general, it may be said that the presence of lime hastens the putrefactive process. This is the reason it is so largely used in making composts.
It is difficult to say just what soils will be improved by liming and what will not, and it is a matter which must be settled by experiment in each case. As a rule heavy clays and loams are benefited, yet of two such soils, apparently identical, one may not be affected in any marked degree while the other may readily respond to treatment. Sandy soils are often improved but sometimes not. Sour, boggy lands, are usually improved by the addition of enough lime to neutralize their undue acidity. Marsh grasses and plants are more tolerant of acid in the soil than tame grasses are, so that in unlimed soil the former run out the latter. The application of lime alone to a very poor soil does not pay.
The particles of lime resting in the soil are partially dissolved by the next rainfall after application, or by the soil moisture, forming lime water, and the lime is distributed in this form through the soil to some extent. It all probably soon becomes converted into carbonate as ground air is usually quite rich in carbon dioxid. Indeed, for many soils, it is immaterial whether lime be applied as lime or as carbonate, granting, of course, that the latter be ground to a fine powder. Economy is in favor of the lime, however, not only because it needs no grinding, but because it is lighter than the corresponding amount of carbonate, making a saving in transportation. The difference is quite considerable, fifty-six pounds of lime being equivalent in effect to 100 pounds of carbonate. For these reasons as well as because it possesses some valuable properties not shared by the carbonate, it is possible that for most localities lime is to be preferred to any form of ground oyster shells, ground limestone, marble dust or the like.
One of these valuable properties not possessed by limestone, is said to be that of acting as a fungicide and insecticide. As a rule, fungi prefer acid reaction in the substances in which they grow, so that the strongly alkaline properties of lime may make a limed soil unsuitable for their growth.[239]
282. Analysis of Lime.—Lime, which is prepared for use as a fertilizer, is rarely submitted to a chemical examination. It is easy to [Pg 306] see, however, that such an examination is of some importance. If the real value of a sample be dependent on the content of lime, the actual quantity present as determined by analysis, must fix the value for agricultural purposes. The more important things to be determined are the quantities of lime, and of slaked lime, of undecomposed calcium carbonate, and of insoluble matter. It will be also of interest to determine the respective quantities of lime present as oxid, hydrate, and carbonate. If any question be raised in the case of slaked lime in respect of its origin, it can usually be answered by an examination of the unburned or unslaked residues. In perfectly slaked lime containing no débris, the analyst will be unable to discover whether it has been made from limestone, marble or shells. The lime used for agricultural purposes should be reasonably free of magnesia, and should not be air-slaked before transportation to the field. In dry air-slaking, a considerable quantity of carbonate may be formed.
283. The Process.—(1) Insoluble and Soluble Constituents.—A representative sample of the lime having been secured, it is reduced to a powder and passed through a half millimeter mesh sieve or ground to a fine powder in an agate mortar. Digest two grams of this sample with an excess of hydrochloric acid, for two hours with frequent stirring; filter, wash the residue with hot distilled water until chlorin is all removed, and dry to constant weight. The lime, magnesia, silica, and other constituents of the filtrate, are determined by the usual processes of mineral analysis.[240]
(2) State of Combination of the Lime.—In a lime containing only small quantities of magnesia the lime carbonate may be determined by estimating the carbon dioxid by any one of the reliable processes in use.[241] In every case sufficient acid must be employed to combine with all the bases present. Tartaric or hydrochloric acid may be used. From the volume or weight of the carbon dioxid obtained the quantity of calcium carbonate may be calculated. Since magnesium carbonate is more easily decomposed by heat than the corresponding calcium compound, any residual carbonate in a well-burned sample is probably lime. The total [Pg 307] percentage of lime in the sample is to be determined in the usual way by precipitation as oxalate and weighing as carbonate or oxid. The lime existing as oxid can be determined by exposing a weighed sample in an atmosphere of aqueous vapor until all the lime is slaked. After drying at 100° the increase in weight is determined and the calcium oxid calculated from the formula, CaO + H₂O = CaO₂H₂.
If now the total lime be represented by a; the lime combined as carbonate by b; and that present as oxid by c; the quantity x existing as hydrate may be calculated from the equation
x = a - (b + c).
| Example: | Let the total lime be | 88 | per | cent. |
| CaO as carbonate, | 2 | “ | “ | |
| CaO as oxid, | 78 | “ | “ |
Then the CaO as hydrate = 88 - (2 + 78) = 8 per cent. The total lime as oxid and hydroxid may also be separated from that present as carbonate by solution in sugar.[242] One gram of calcium oxid is completely soluble in 150 cubic centimeters of a ten per cent sucrose solution. Magnesia, iron and alumina do not interfere with the determinations.
284. Gypsum or Land Plaster.—This substance is highly prized as a top dressing for grass and for admixture with stall manure for the purpose of fixing ammonia. Its value in both cases depends upon its percentage of hydrated calcium sulfate. The quantity of gypsum mined in the United States in 1893 was a little over 250,000 tons. Of this amount only about 50,000 tons were used as fertilizer.[243] In the same time there were imported into the United States, in round numbers, 170,000 tons. If the same proportionate part of this were used for fertilizing purposes, it may be said that the annual consumption of land plaster in the United States at the present time for agricultural uses is about 75,000 tons.
Gypsum, being a very soft mineral, is easily ground and should be in the state of a fine powder when used for fertilizing purposes. It is soluble in about 500 parts of rain water, so that when applied as a top dressing it is carried into the soil by rain. Its favorable action is both as a plant food and mechanically in modifying, in an advantageous way, the physical constituents of the soil. It is also valuable for [Pg 308] composting and for use in stables by reason of its power of fixing ammonia by the formation of lime carbonate and ammonium sulfate:
(H₄N)₂CO₃ + CaSO₄ = (H₄N)₂SO₄ + CaCO₃.
285. Analysis of Gypsum.—For agricultural purposes it will be sufficient to determine the quantity of sulfuric acid, and to calculate therefrom the amount of calcium sulfate in the sample: Or the lime may be determined and the quantity of sulfate calculated therefrom.
(1) Insoluble Matter.—In the conduct of the work the sample of gypsum is rubbed to an impalpable powder in an agate mortar. The washed sample, about one gram, is dissolved in a large excess of dilute hydrochloric acid, the digestion being continued at near the boiling-point, with frequent stirring, for at least two hours. The solution is made alkaline, filtered, and the residue washed and dried to constant weight.
(2) Sulfuric Acid.—The washings and filtrate from the above determination are made up to a definite volume with water and divided into two equal parts. The sulfuric acid is estimated in one part by adding to it sodium carbonate until the acidity is nearly neutralized. The sulfuric acid is then thrown down at near boiling temperature by the gradual addition of barium chlorid solution. The barium sulfate formed is separated, washed, dried, and weighed in the usual manner.
(3) Iron and Alumina.—To the other half of the solution a little nitric acid is added and boiled to convert any ferrous into ferric iron. On the addition of ammonia the iron and alumina are separated as hydroxids, collected on a gooch, washed, dried, ignited, and weighed as oxids.
(4) In the filtrate the lime is thrown out as oxalate, and separated and weighed in the usual way as oxid. One part of CaO is equal to 2.4286 parts of CaSO₄.
(5) Moisture.—Dry about two grams of the sample to constant weight at 80°.
(6) Water of Crystallization.—Heat the residue from the above to 150°, until a constant weight is obtained. The loss represents water of crystallization.
(7) Carbonates.—Determine the quantity of carbon dioxid evolved [Pg 309] by the usual process, and calculate the calcium carbonate.
286. Solution in Sodium Carbonate.—Gypsum is also easily decomposed by boiling with a solution of about ten times its weight of sodium carbonate. The calcium, by this operation, is converted into carbonate and can be collected on a gooch, washed, and estimated as usual, but in this case it will contain all the insoluble matters, from which the lime can be separated by solution in hydrochloric acid.
In the filtrate from the above separation the excess of sodium carbonate is removed by the addition of hydrochloric to slight acidity, and the sulfuric acid estimated as described in the preceding paragraph.
Pure gypsum has a composition represented by the following formula: CaSO₄·H₂O.
It contains:
| Sulfur dioxid | 46.51 | per | cent. |
| Lime | 32.56 | “ | “ |
| Water | 20.93 | “ | “ |
A commercial sample of ordinary gypsum should have about the following composition:[244]
| CaSO₄·H₂O | 88.15 | per | cent. |
| CaCO₃ | 3.50 | “ | “ |
| Fe₂O₃ and Al₂O₃ | 1.50 | “ | “ |
| Insoluble | 2.80 | “ | “ |
| Organic matter | 0.50 | “ | “ |
| Water and undetermined | 3.55 | “ | “ |
287. Common Salt.—Common salt is highly esteemed in many quarters as a top dressing for lawns and meadows, and also for cultivated crops. Its action is chiefly of a mechanical and katalytic nature, since it does not form a very large percentage of the mineral food of plants. On account of its affinity for moisture it is also said to have some value as a condenser and carrier of water in times of drouth. On account of its great cheapness, selling often for less than ten dollars a ton, its use in moderate quantity entails no great expense. Its ability, however, to pay for its own use in the increased harvest is of a doubtful character when it is applied at a cost of more than a few dollars per acre. In the chemical examination of a sample of [Pg 310] common salt which is to be used as a fertilizer, a complete analysis is rarely necessary. When desired it can be conducted according to the usual methods of mineral analysis. For practical purposes the moisture, insoluble matter, magnesia and chlorin should be determined and the quantity of sodium chlorid calculated from the latter number. Traces of iodin or bromin which may be present are of no consequence.
The moisture is determined by drying two grams of the well-mixed and finely-powdered sample to constant weight at 100°. The chlorin is obtained by precipitation of an aliquot part of a solution of the salt by set silver nitrate, using potassium chromate as indicator.
In the determination of insoluble matter it should not be forgotten that a little gypsum may be present, and this should be dissolved by rubbing to a finer powder and by repeated digestion in water. The magnesia and lime are separated and determined in the usual manner. If the quantity of gypsum present be sufficient to warrant it the sulfuric acid may be separated and weighed in the manner already described. Common salt when present in the soil in proportions greater than one-tenth per cent is injurious to vegetation.
288. Green Vitriol.—When iron is used as a fertilizer it is usually applied as ferrous sulfate. The value of iron in a soil is incontestable and by reason of the fact that fertile soils are always well aerated the iron present in the arable layer is found in the ferric state. When green vitriol is applied to the soil it undergoes gradual oxidation and appears finally in a more highly oxidized form. Iron acts directly on the plant in promoting the development of the chlorophyll cells, and is also found in almost all parts of the vegetable organism. A too great quantity of ferrous sulfate is destructive of plant growth in which respect it resembles common salt. It should therefore be applied with due regard to the dangers which might arise from an excessive quantity. It is not likely, however, that when applied in a finely powdered state at the rate of from two to four hundred pounds per acre it would ever prove poisonous to vegetation.
In the analysis of a sample of green vitriol it will be sufficient to determine the moisture, water of crystallization, iron, and sulfuric [Pg 311] acid. The moisture may be ascertained by drying the finely powdered sample over sulfuric acid for a few hours. The water of crystallization is separated by exposing the sample to a temperature of 285° for two hours. The iron may be determined by oxidizing to the ferrous state by boiling with nitric acid and then precipitating with ammonia, and proceeding as directed for iron analysis. The sulfuric acid is separated as barium sulfate and determined as already directed.
289. Stall Manures.—There are no definite methods to be described for the analyses of that large class of valuable fertilizer produced in the stable and pen, and which collectively may be called stall manures. The methods of sampling have already been described,[245] but only patience and tact will enable the collector to get a fair representation of the whole mass. These manures are a mixture of urine, excrement, waste fragments of fodder, and the bedding used for the animals. With them may also be included the night soil and waste from human habitations and the garbage from cities. All of these bodies contain valuable plant foods and the phosphoric acid, potash, and nitrogen therein are to be determined by the methods already given for these bodies when they occur in, or are mixed with, organic matter. In general, stall manures are found to have a higher manurial value than is indicated by the amount of phosphorus, potash, and nitrogen which they contain. Through them there is introduced into the soil large quantities of humus bodies whereby the physical state of the soil is profoundly modified and its adaptability to the growth of crops, as a rule increased. The addition of active nitrifying ferments in stall manure is also advantageous. Stall manures, however, may in many cases prove to be injurious to a crop, as for instance, when they are applied in a poorly decomposed state and in a season deficient in moisture.
It is essential therefore that the bedding of animals be in a finely divided state, whereby not only are the absorptive powers of the organic matter increased but also the conditions for their speedy decay favored. To avoid the loss of ammonia arising from decomposing urine it is advisable to compost the stall manure with gypsum or to sprinkle it from time to time with oil of vitriol. [Pg 312]
In the analysis the moisture may be estimated by drying a weighed portion of the sample to constant weight at 100° or at a lower temperature in a vacuum. The potash and phosphoric acid are determined as usual, with previous careful incineration, and the nitrogen secured by the moist combustion process.
290. Hen Manure.—This fertilizing substance is a mixture of the excrement of the fowl yard with feathers, dust, and other débris. Measured by the standard applied to commercial fertilizers hen manure has a low value. As in the case of other farm manures, however, it produces effects quite out of proportion to the amount of ordinary plant foods which it contains. In a sample examined at the Connecticut station the percentages of fertilizing constituents were found to be the following[246]:
| Water | 51.84 |
| Organic and volatile matters | 24.27 |
| Ash | 23.89 |
The organic matter contained 0.61 per cent of nitrogen as ammonia and the ash 0.97 per cent of phosphoric acid, and 0.59 per cent of potash, all calculated to the original weight of the sample. The percentage of water in this sample is undoubtedly higher than the average, so that it can hardly be taken to represent the true composition of this manure. The potash, phosphoric acid, and nitrogen are to be determined by some one of the standard methods already described, the two former after careful incineration.
291. Guanos and Cave Deposits.—The principal constituents of value in these deposits are nitrogen and phosphoric acid. The other organic matters are also of some value but have no commercial rating. The nitrogen may be present in all its forms; viz., organic, ammoniacal, amid, and nitric, and for this reason is well suited not only to supply nourishment to the plant in the earlier stages of its growth but also to cater to its later wants. In guano deposits in caves, due usually to the presence of bats, similar forms of fertilizers are found and the soluble constituents due to decay and nitrification are protected from the leaching to which they would be subjected in the open air.
In many localities in the United States these deposits are found, but the humidity of our climate has prevented the immense open deposits of [Pg 313] guano that characterize some of the arid islands of the Pacific Ocean.
Many bat guanos examined in this laboratory have also been found to contain potash, in one case 1.78 per cent. It is suggested therefore that the analyst do not omit to examine each sample qualitatively for this substance and to determine its amount when indications point to its presence in weighable proportions. In the many samples of bat guano of American origin which have been analyzed in this laboratory in the last few years some very rich in plant food have been found. In one instance the total percentage of nitrogen present, was 10.11 per cent. In some cases the phosphoric acid is high but rarely in conjunction with a high content of nitrogen. In one instance where the total phosphoric acid reached 14.53 per cent, the content of nitrogen was 4.87 per cent.
In respect of the processes of analysis there are no especial directions to be given. The phosphoric acid, as given below, and the potash are to be determined by the usual methods, the total phosphoric acid and potash after the destruction of the organic matter.
In old cave deposits the processes of decay and nitrification seem to have long been completed and we have found very little power of inducing nitrification in culture solutions seeded from these samples.
292. French Official Method for Total Phosphoric Acid in Guanos.—To determine the phosphoric acid in guanos, the method officially adopted by the French agricultural chemists may be used.[247]
Two grams of the sample are rubbed up in a porcelain crucible with a decigram of slaked lime to prevent the possible reduction of the phosphoric acid by the organic matter. The mixture is slightly moistened with a few drops of water, dried on a sand-bath, and afterwards heated to redness, best in a muffle, until organic matter is destroyed. The contents of the crucible are detached and placed in a flask of 200 cubic centimeters capacity. The crucible is well digested twice with some hydrochloric acid to dissolve any adhering fragments, and finally washed with hot water, the acid and water being added to [Pg 314] the flask. The contents of the flask are boiled for fifteen minutes and then poured into a flat-bottomed dish, the flask well rinsed three or four times with small quantities of water, and the liquor and washings are evaporated to dryness to render the silica insoluble. The residue is taken up by a mixture of ten cubic centimeters each of hydrochloric acid and water, heated for a few minutes and filtered, and the dish well washed with successive small portions of water, but the total volume of the filtrate and washings should not exceed eighty cubic centimeters. In this filtrate the phosphoric acid may be determined by any one of the approved methods.
293. Waste Leather.—This material belongs probably to that class of nitrogenous substances which has already been considered in paragraph 149. The chief manurial value of the waste is found in its nitrogenous content. The value of this for available plant food has been investigated by Lindsey.[248] A complete resumé of the literature of the subject is also given by him.
The best way of identifying leather waste is by the process proposed by Dabney.[249] It depends on the color produced in a solution of iron phosphate by the tannin compounds derived from the leather. The reagent is prepared by dissolving a freshly made precipitate of iron phosphate from ten grams of ferric chlorid in 400 cubic centimeters of an aqueous solution of forty grams of glacial phosphoric acid. A gentle heat promotes the solution of the phosphate.
In the case of a fertilizer supposed to contain leather, about one gram of the material is treated with thirty cubic centimeters of water and a few drops of sulfuric acid. The mixture is boiled and poured on a filter. To a portion of the filtrate some of the solution of iron phosphate is added, and the mixture made alkaline with ammonia. If leather be present in the sample, a purple or wine color will be developed. Lindsey could easily detect the leather when it was added in ten per cent quantities by the above method, and he regards this method as superior to the microscope which is unreliable in the case of finely ground material.
While leather, as such, decays slowly, and therefore is not at once available for the nourishment of plants it acquires greater utility [Pg 315] after digestion in sulfuric acid. Artificial digestion experiments with leather previously treated with sulfuric acid show that, approximately, seventy per cent of the nitrogen pass into solution. Such a prepared leather has, therefore, a digestive coefficient in respect of nitrogen not much inferior to most organic bodies.
In comparative trials with sodium nitrate it was demonstrated that nitrogen in leather, previously dissolved in sulfuric acid, has a rank of about sixty when it is rated at one hundred in the soda salt.
For the estimation of the nitrogen in leather the moist combustion process is to be preferred.
294. Analysis of Wood Ashes.—The only kinds of ashes used extensively for manurial purposes are those derived from the burning of hard woods. The ash of soft woods, such as the pine, is too poor in plant foods to warrant its transportation to any great distance for manurial purposes. The methods of incineration of organic bodies for the purpose of obtaining and estimating their mineral contents will be fully discussed in the third volume of this work.
It is important in ash analysis to know whether there be enough of iron present to combine with all the phosphoric acid. For manurial purposes it will be found sufficient to determine the percentages of potash and phosphoric acid alone. For hygienic purposes it is advisable to examine the ash qualitatively and, if necessary, quantitatively for zinc, lead, copper, boric acid, and other bodies of a similar character which may be naturally present in the ash, or may have been added to the organic substance from which it was prepared for preservation or other purposes. The methods of making these special investigations will be discussed in the succeeding volume. At present will be given, however, not only the methods for detecting phosphoric acid and potash, but also for a complete analysis of an ash in so far as its usual constituents are concerned.
295. Carbon, Sand, and Silica.—The official agricultural chemists have recommended the following procedure for the determination of the unburned carbon, and the sand and silica.[250]
Five grams of the ash are treated in a beaker, covered with a [Pg 316] watch glass with fifty cubic centimeters of hydrochloric acid of 1.115 specific gravity, and digested on the water-bath until all effervescence has ceased. The cover is then removed and the liquid evaporated to complete dryness to render the silica insoluble. The residue is moistened with two or three cubic centimeters of hydrochloric acid and taken up with about fifty cubic centimeters of water, allowed to stand on the water-bath a few minutes, filtered, and thoroughly washed. The filtrate and washings are made up to a quarter of a liter for analysis. The residue is washed from the filter into a platinum dish and boiled about five minutes with twenty cubic centimeters of a saturated solution of pure sodium carbonate; afterwards a few drops of pure sodium hydroxid solution are added and the liquid allowed to settle, and it is then decanted through a tared gooch. The residue is boiled with sodium carbonate solution and decanted as before, a second and a third time, and finally brought upon the felt and thoroughly washed, first with hot water, then with a little dilute hydrochloric acid, and finally with hot water until free of chlorids. The residue in the gooch is dried at 110° to constant weight, giving the carbon and sand. It is then incinerated and the weight of the sand determined, the difference giving the carbon. It is advisable to examine the sand with a microscope to determine if it be pure. The alkaline filtrate and washings from the carbon and sand are acidified with hydrochloric, evaporated to dryness, and the silica separated and determined in the usual way.
Instead of determining soluble silica directly from the sodium carbonate solution, as above, another portion of the ash may be treated with hydrochloric acid and evaporated to dryness as before described, filtered on an ordinary filter, washed, burned, and weighed, giving the weight of silica plus sand, from which the weight of sand is deducted to obtain soluble silica. It is inadmissible to separate the soluble silica from the residue after it has been ignited.
Instead of limiting the quantity of hydrochloric acid used for moistening the dried residue, as suggested above by the official chemists, enough should be employed to fully saturate the mass. The weight of pure ash is obtained by subtracting from the weight of the [Pg 317] sample taken the sum of the weights of carbon, sand, and carbon dioxid.
296. Ferric Phosphate and the Alkaline Earths.—The ferric phosphate, lime, magnesia, and manganese are determined in an aliquot part of the first hydrochloric acid solution and washings obtained above. Fifty or one hundred cubic centimeters may be used, corresponding to one or two grams of the original ash. The accurately measured quantity of the solution is carefully treated with ammonia until the precipitate formed on its addition becomes permanent on shaking. Ammonium acetate and acetic acid are then added until the mixture has assumed a strongly acid reaction. The separation of the ferric phosphate precipitate is promoted by gentle warming, and it is separated by filtration without unnecessary delay. If the precipitate be not large the sample contains no manganese and alumina in weighable quantities, and if the filtrate be not red the precipitate be washed with hot water containing a little ammonium nitrate. It is then ignited and weighed as Fe₂P₂O₈ and the quantity of ferric oxid computed therefrom. If, however, the precipitate be large it is well washed as above and then dissolved in as small a quantity as possible of hydrochloric acid, and the solution is again precipitated as above by the addition of ammonia, ammonium acetate, and acetic acid. The ferric phosphate obtained by the second precipitation is treated exactly as above described.
In case, however, any weighable quantities of manganese or alumina are present it will not do to weigh the precipitate of ferric phosphate directly even after a second precipitation. Also if the filtrate at first obtained have a red color the precipitate may contain basic ferric phosphate. In this latter case it should be ignited and weighed, then dissolved in hydrochloric acid and the ferric oxid estimated in the solution and from the difference the quantity of combined phosphoric acid calculated.
The separation of the iron from the phosphoric acid may be accomplished by adding tartaric acid to the hydrochloric acid solution of the iron phosphate above obtained and then ammonium chlorid and ammonia. The mixture is placed in a flask and ammonium sulfid added. The flask is [Pg 318] closed, placed in a warm place, and allowed to stand until the supernatant liquid is clear and of a pure yellow color without a trace of green. The iron is separated by filtration, washed, dissolved, and estimated in the usual way.
If manganese and alumina be present the iron and manganese are separated from the phosphoric acid and alumina by the processes just given for the separation of iron from phosphoric acid. In the filtrate the alumina and phosphoric acid are separated as follows: The filtrate is evaporated in a platinum dish after the addition of an excess of pure sodium carbonate until no ammonia is set free by a further addition of the carbonate. Some nitric acid is then added and the evaporation continued to dryness. The residue is fused and after cooling softened with water, washed into a small beaker, some hydrochloric acid added, warmed, and filtered. Ammonia is next added until the reaction is alkaline. If no precipitate be produced no alumina is present. In this case more nitric acid is added, the solution again evaporated, and the phosphoric acid determined by the usual methods.
In case a precipitate is formed, showing the presence of alumina, nitric acid is added until the precipitate is dissolved, and then in slight excess and after evaporation the phosphoric acid separated by molybdic solution and determined as usual. From the filtrate the excess of molybdic acid is removed by hydrogen sulfid and the alumina determined in the filtrate: Or the alumina may be determined directly in the hydrochloric acid solution of the melt above obtained as aluminum phosphate by adding sodium phosphate, ammonia and acetic acid. The aluminum phosphate is separated by filtration and determined in the usual manner. The phosphoric acid is then determined in another aliquot part of the original filtrate from the first solution of the ash.
Since most ashes contain an excess of phosphoric acid above the quantity required to combine with the iron it is preferable to proceed on that basis as described in the next paragraph.
297. Method Used in this Laboratory.—The principle of the method rests on the assumption that all the phosphoric acid may be removed from the solution by the careful addition of iron chlorid. Any [Pg 319] excess of iron is then removed by ammonium acetate and the manganese, lime, and magnesia, are separated in the filtrate. The percentage of iron is determined by reduction of the iron in another portion of the solution and titration with potassium permanganate. The process as conducted by McElroy, is as follows:[251]
Moisture.—If the ash contain much carbon the water is best determined by drying in vacuo to avoid oxidation.
Sand, Silica, and Carbon.—Place a portion of the ash in a weighed platinum dish, weigh, and cover the sample with hydrochloric acid of 1.115 specific gravity. Evaporate to dryness on the water-bath, and then heat for fifteen minutes at 105° to 110° in an air-bath. Repeat the treatment with acid and drying: Finally cover with a third portion of acid and digest on the water-bath for an hour or two. Filter into a weighed gooch and wash the residue free of chlorids. The gooch is best weighed with the dish to avoid the necessity of transferring the silica which may adhere to the sides of the former. Dry at a few degrees above the boiling temperature of water and weigh. Where the ash contains much charcoal the drying is best done in a vacuum at 60° to 70°. The increase in weight found represents sand, silica, and carbon: Burn and reweigh. The loss is carbon.
Another portion of ash is treated as before except that it is filtered through a paper filter. The filtrate is united with that of the previous sample in a graduated flask. When the washing is completed the filter is placed in the dish, a weak solution of caustic soda added, and the mixture heated on the water-bath for some time. Decant while hot through a fresh filter and retreat the residue in the dish with another portion of alkali. Finally wash with hot water till the alkaline reaction disappears, then with weak hydrochloric acid, then with water until chlorids disappear. The washed mass on the filter is transferred to a platinum dish and ignited. The weight obtained represents sand.
Separation of Phosphoric Acid.—The united filtrates from the two determinations are placed in a graduated flask and made up to the mark. An aliquot portion of this solution representing half a gram of [Pg 320] the original ash or any other convenient quantity is transferred to a beaker and a solution of ferric chlorid added until ammonia produces a brown precipitate in the mixture. Neutralize with ammonia and hydrochloric acid alternately until the liquid is as little acid as it can be and still remain clear. Add from ten to twenty cubic centimeters of a solution of sodium acetate (1:10) and bring to a boil. The liquid should be quite dilute. Filter and wash free of chlorids with boiling water containing some sodium acetate.
Manganese.—Make the filtrate faintly alkaline with ammonia and add ammonium sulfid. Any manganese sulfid which may form is separated by filtration, treated with dilute acetic acid and the resulting solution, which should be clear, heated to boiling, nearly neutralized with caustic soda, and mixed with bromin water. The resultant manganese dioxid is to be filtered into a gooch, ignited and weighed as Mn₃O₄.
Lime.—Reacidify the filtrate from the manganese sulfid with acetic acid, heat to boiling and add ammonium oxalate: Allow to stand over night, filter through a gooch and wash with water containing acetic acid. The calcium oxalate can be weighed as such, but it is preferable to dry thoroughly and then heat in a small bunsen flame until a change can be noted passing over the precipitate. If this is carefully done the residue will be calcium carbonate. In any case the result is to be checked by igniting over the blast lamp to constant weight and weighing the lime thus obtained.
Magnesia.—In the filtrate the magnesia can be determined by sodium phosphate in the usual manner. In very accurate work the calcium oxalate obtained as directed above can be dissolved and reprecipitated, and the magnesia in the filtrate added to that in the first filtrate.
Iron.—For iron another aliquot portion of the original solution is taken, acidified with sulfuric evaporated to drive off hydrochloric acid, rediluted and passed through the Jones reductor described in paragraph 112. The filtrate is titrated with potassium permanganate solution in the usual manner.
Alkalies.—For the alkalies another aliquot portion is taken and precipitated while hot, with barium chlorid and barium hydrate, filtered, and ammonia and ammonium carbonate added to remove the excess [Pg 321] of barium salt. Refilter, evaporate to dryness in a platinum dish, and ignite gently to expel all ammonia salts, repeat this operation after taking up with water and finally heat to constant weight. The weight obtained represents a mixture of potassium and sodium chlorids, with usually carbon derived from impurities in the ammonia. A little magnesia is often present. The potassium is estimated by means of platinum solution, and the potassium chlorid found deducted from the total weight gives the sodium chlorid. The carbon is usually unweighable, though it often looks as if present in considerable quantity. It may be estimated, however, by dissolving the mixed chlorids in weak hydrochloric acid and filtering through a gooch before making the potassium estimation. The estimation of the magnesia remaining with the mixed chlorids may be effected by evaporating the alcoholic solution remaining after the precipitation of the potassium to dryness, redissolving in water, placing the solution in a flask provided with gas tubulures, introducing hydrogen, and placing in the sunlight. The platinum is soon reduced, leaving the liquid colorless. Heating facilitates the reaction. Displace the hydrogen by a current of carbon dioxid, filter, concentrate the solution and precipitate the magnesia by sodium phosphate in the usual manner.
Phosphoric Acid.—It is best to determine the phosphoric acid directly in an aliquot part of the first filtrate from the hydrochloric acid solution of the ash obtained as described under the determinations of sand, silica and carbon. When there is not enough of the material for this, the precipitate of ferric phosphate may be dissolved and the phosphoric acid determined after separation with ammonium molybdate.
Sulfuric Acid.—Fifty cubic centimeters of the original hydrochloric acid filtrate, obtained as described under the determinations of sand, silica and carbon, are heated to boiling, and the sulfuric acid thrown out by the gradual addition of barium chlorid. During the precipitation the mixture is kept at the boiling temperature, but taken from the lamp and the precipitate allowed to settle from time to time until it is seen that an additional drop of [Pg 322] the reagent causes no further precipitate. The barium sulfate is collected, dried, and weighed in the usual manner.
Chlorin.—Dissolve from one to five grams of the ash in nitric acid in very slight excess, or in water. If the solution be made in nitric acid the excess must be neutralized if the chlorin be determined volumetrically; and if the solution be in water, nitric acid must be added if the determination be gravimetric.
The volumetric determination is accomplished in the usual manner with a standard silver nitrate solution, using potassium chromate as indicator. The gravimetric determination is effected by precipitation with silver nitrate, collecting, washing, and drying at 150° the silver chlorid obtained.
Carbon Dioxid.—The carbon dioxid is most conveniently estimated in from one to five grams of the ash, according to its richness in carbonates, by the apparatus described in volume first, or some similar device.[252]
298. Official Method for Determinations of the Alkalies.—Evaporate the filtrate and washings from the sulfuric acid determination, paragraph 297, in a porcelain dish to dryness, redissolve in about fifty cubic centimeters of water and add milk of lime, or barium hydroxid solution, which must be perfectly free from alkalies, until no further precipitation is produced, and it is evident there is an excess of calcium hydroxid or barium hydroxid present; boil for two or three minutes, filter hot, and wash thoroughly with boiling water, precipitate the lime and baryta from the filtrate with ammonia and ammonium carbonate, filter, evaporate the filtrate to dryness in a porcelain dish, and drive off the ammonia salts by heat below redness.[253] When cold, redissolve in fifteen or twenty cubic centimeters of water, precipitate again with a few drops of ammonia and ammonium carbonate solution, let stand a few minutes on the water-bath and filter into a tared platinum dish and evaporate to dryness, expel the ammonia salts by heating to just perceptible dull redness, weigh the potassium and sodium chlorids obtained and determine the potassium chlorid with platinic chlorid as usual.
The potassium may also be determined by the perchlorate method, or the total chlorin be determined volumetrically, and the relative percentages of potassium and sodium chlorids calculated by the usual [Pg 323] formula: Or multiply the weight of chlorin in the mixture by 2.1035, deduct from the product the total weight of the chlorids and multiply the remainder by 3.6358. The product expresses the weight of the sodium chlorid contained in the mixed salts. The indirect method is only applicable when there are considerable quantities of alkalies present and where they exist in approximately molecular proportions. It is therefore a process rarely to be recommended in ash analysis.
299. Statement of Results.—The bases which are found present in the ashes of wood and other vegetable tissues exist without doubt before incineration, chiefly in combination with inorganic acids. Even the phosphorus and sulfur which after ignition appear as phosphates and sulfates, have previous thereto existed in an organic form to a large extent. The silica itself is profoundly modified in the organism of the growing plant and doubtless does not exist there in the purely mineral form in which it is found in the ash. During the progress of incineration, with proper precautions, all the phosphorus and sulfur are oxidized and appear as phosphoric and sulfuric acids. The silica is reduced to a mineral state, and if a high heat be employed silicates are formed. The organic salts of lime, magnesia and other bases at a low temperature are converted into carbonates, and if a higher temperature be used, may appear as oxids. The organic compounds of alkalies will be found in the ash as carbonates. It would be useless, therefore, to try to state the results of ash analysis in forms of combination similar to those existing in the original vegetable tissues. It is not certain even that we can in all cases judge of the form of combination in which the different constituents exist in the ash itself. It is therefore to be preferred in a statement of ash analysis to give the bases in the form of oxids, and the sulfur and phosphorus in the form of anhydrids, and the chlorin in its elementary state. In this case an equivalent amount of oxygen to the chlorin found must be subtracted from the total. If an attempt be made to combine the acid and basic elements the chlorin should first be united with sodium, and any excess thereof with potassium, and the amount of base so combined calculated to oxid and deducted from the total of such base or bases present. The carbonic acid present should be combined first [Pg 324] with alkalies after the chlorin has been supplied. The phosphoric acid should be combined first with the iron and afterwards with lime or magnesia. In all cases the percentages should be based upon the ash, after the carbon and sand have been deducted, or it is also convenient at times to throw out of the results the carbon dioxid and to calculate the other constituents to the ash free of that substance. In determining the quantities of mineral matters removed from soil by crops, the ash should be determined with great care, freed of carbon and sand, and the calculations made on the percentage thus secured. In all statements of percentages of the essential constituents of ash, as regards fertilizing materials, it should be specified whether the percentage is calculated on a crude ash, the pure ash, that is free of carbon and sand, or upon a basis excluding the carbon dioxid. For the purpose of fertilizer control, the analyst and dealer will be satisfied, as a rule, with the determination of the percentages of phosphoric acid and potash alone. To the other constituents of an ash is not assigned any commercial value.
[239] Compiled for author by Mr. K. P. McElroy.
[240] Volume First, pp. 356 et seq.
[241] Volume First, pp. 337 and 390.
[242] Volume Second, p. 81.
[243] Day: Mineral Resources of the United States, 1893, p. 713.
[244] Frankland: Agricultural Analysis, p. 240.
[245] Volume Second, p. 9.
[246] Annual Report Connecticut Agricultural Experiment Station, 1888, Part 1, p. 80.
[247] Rapport addressé par le Comté des Stations Agronomiques, 1887, p. 34.
[248] Agricultural Science, Vol. 8, pp. 2 and 3, 1894, and Massachusetts Agricultural Station, 12th Annual Report, pp. 285 et seq.
[249] North Carolina Agricultural Experiment Station, Bulletin No. 3.
[250] Bulletin No. 43, Chemical Division, U. S. Department of Agriculture, p. 390.
[251] Compiled for author by Mr. K. P. McElroy.
[252] Volume First, p. 337.
[253] Vid op. cit. 12, p. 391.
[Pg 325]
Transcriber’s Notes:
Old spellings were not corrected, e.g "dioxid" insread of "dioxide".
The illustrations have been moved so that they do not break up paragraphs and so that they are next to the text they illustrate.
Typographical and punctuation errors have been silently corrected.